
Case Report
Austin J Endocrinol Diabetes. 2019; 6(2): 1071.
A Case of Partial Nephrogenic Diabetes Insipidus Associated with Anidulafungin Treatment
Sarma S1*, Bahrami J2, Martin L3 and McInnes N4
1Endocrinology and Metabolism, University of Toronto, Canada
2Endocrinology and Metabolism, McMaster University, Canada
3Division of General Internal Medicine, McMaster University, Canada
4Division of Endocrinology and Metabolism, McMaster University, Canada
*Corresponding author: Sarma S, Department of Medicine, University of Toronto.190 Elizabeth Street, Toronto, ON M5G 2C4. Canada
Received: November 28, 2019; Accepted: December 24, 2019; Published: December 31, 2019
Abstract
Nephrogenic Diabetes Insipidus (NDI) is classically associated with lithium, however it can be caused by a number of drugs including the anti-fungal agent amphotericin B. Here we describe a case of a 52-year-old male presenting with NDI following treatment with anidulafungin. The patient was admitted with a strangulated ventral hernia complicated by post-operative sepsis for which anidulafungin was initiated. Ten days after initiation of anidulafungin, his plasma sodium level rose to 167 mmol/L with serum and urine osmolality of 357 and 436 mmol/kg, respectively. Anidulafungin was discontinued and 5% dextrose in water (D5W) and vasopressin infusions were initiated. There was a partial response to the vasopressin infusion, suggestive of NDI. Sodium levels eventually returned to normal with continued vasopressin, and later intravenous desmopressin and D5W infusions. The patient received no other medications associated with NDI. Changes in sodium, and serum and urine osmolality, as well as delayed resolution of NDI fit with the fifty-hour half-life of anidulafungin. This case suggests that anidulafungin may be associated with NDI.
Keywords: Diabetes insipidus; Anidulafungin; Nephrogenic diabetes insipidus; Echinocandin; Antifungal
Abbreviations
ADH: Anti-Diuretic Hormone; NDI: Nephrogenic Diabetes Insipidus; CT: Computed Tomography; IV: Intravenous; PEA: Pulseless Electrical Activity; ICU: Intensive Care Unit; D5W: Dextrose 5% in Water; FDA: Food and Drug Administration
Introduction
The osmoregulatory system maintains plasma sodium within a narrow range (135-142 mmol/L) by controlling free water intake and loss [1]. The differential diagnosis for hypernatremia includes (i) unreplaced free water loss, (ii) severe metabolic stress (such as severe exercise or seizures) leading to water entry into cells, or (iii) sodium overload through administration of hypertonic solutions or salt poisoning [1]. Unreplaced free water loss can be caused by insensible or gastrointestinal losses, diabetes insipidus, or osmotic diuresis. Lack of access to free water or an impaired thirst mechanism can lead to hypernatremia. The physiologic response to hypernatremia with free water loss is to maximally concentrate urine. Diabetes insipidus is defined by an impaired ability to concentrate urine, resulting in polyuria with hypotonic urine and serum hypernatremia [2]. This occurs due to central diabetes insipidus, from an inability to produce ADH; NDI, due to inability of nephrons to respond to circulating ADH; or gestational diabetes insipidus, which is due to rapid metabolism of circulating ADH [3]. Response to exogenous ADH (desmopressin) is necessary to differentiate central from nephrogenic types [2].
Medications are the most common cause of acquired NDI in adult patients. Lithium impairs the ability to concentrate urine starting at eight weeks after initiation [4,5]. Other drugs associated with NDI include the anti-viral agents foscarnet [6] and cidofovir [7] and the anti-fungal agent amphotericin B [8]. Demeclocycline, an antibiotic in the tetracycline class, has been associated with NDI in several case reports [9].
Metabolic causes of NDI include hypercalcemia and hypokalemia. Hypercalcemia can cause calcium deposits in the distal tubules and collecting ducts, leading to decreased expression of aquaporin-2 channels [10]. Causes of chronic renal failure associated with NDI include sickle cell anemia, polycystic and medullary kidney disease [11-13], renal amyloidosis, and Sjogren’s syndrome [14]. Postobstructive and osmotic diuresis associated with NDI occurs due to down-regulation of aquaporin-2 channels [15].
Here we present a case of a patient who experienced partial NDI following treatment with the anti-fungal agent anidulafungin.
Case Presentation
A 52-year-old man presented to the emergency department with a three-day history of abdominal pain, obstipation, and fever. His medical history included obesity, recurrent leg ulcers, and a small bowel resection. His home medication was ibuprofen as needed for pain. Abdominal radiographs showed multiple air fluid levels, and subsequent CT scan with Intravenous (IV) contrast confirmed small bowel obstruction and a perforated ventral hernia. The patient was started on IV piperacillin-tazobactam, cefazolin, and lactated ringer’s IV fluid for hypotension. He underwent laparotomy and small bowel resection with hernia repair. He experienced new onset atrial fibrillation during surgery, resulting in a PEA arrest and needed resuscitation with epinephrine and norepinephrine. Atrial fibrillation was managed with metoprolol, amiodarone, and digoxin. A noncontrast CT head for decreased level of consciousness showed no intracranial abnormalities. He was taken to the ICU post-operatively and underwent a second look laparotomy with bowel anastomosis.
The patient had post-operative fevers and blood cultures grew Klebsiella oxytoca and abdominal fluid culture grew Escherichia coli. Piperacillin-tazobactam was switched to meropenem. Due to hemodynamic instability on day 6 of admission, empiric anidulafungin was added. He continued to receive his cardiac medications along with propofol, fentanyl, hydromorphone, midazolam, ondansetron, subcutaneous heparin, and chlorhexidine spray. The patient’s plasma sodium began to rise on day 7 of admission. The sodium peaked at 167mmo1/L on day 16 with concurrent serum and urine osmolality of 357 and 436mmo1/kg, respectively. The serum glucose, potassium, and corrected calcium at the time were normal at 6.4mmo1/L, 3.8mmo1/L, and 2.5mmo1/L, respectively. The renal function had not changed significantly with creatinine at 228umo1/L on the day of admission and 233umo1/L on day 16. Urea was 14.9mmo1/L on day 2 and 15.4mmol/L on day 26. Given the degree of hypernatremia and serum hyper osmolality, normal response to ADH was expected to increase urine osmolality above 500mmol/kg. Inability to fully concentrate urine in the setting of hypernatremia led to the diagnosis of diabetes insipidus.
Anidulafungin was stopped on day 16 due to possible contribution to diabetes insipidus. In addition, D5W and vasopressin infusions were initiated to target a decline in sodium of 10mmol/L per 24 hours. Despite increasing vasopressin to a maximum dose of 2.5 units/hr and administering D5W at 250 mL/hour on day 19 of admission, sodium correction was minimal to 161 mmol/L and urine osmolality only rose to 505 mmol/kg with serum osmolality of 360 mmol/kg. With hyperosmolality, hypernatremia, and vasopressin administration, urine osmolality was expected to be maximally concentrated above 800mmol/kg if ADH response was normal. Therefore, a diagnosis of partial NDI was made.
With ongoing vasopressin and D5W infusions and discontinuation of anidulafungin, his sodium levels returned to 144 mmol/L) by day 22 of admission. The patient was switched to IV desmopressin at 1 mcg twice daily on day 23, which was continued until day 26 to prevent recurrence of hypernatremia. The patient eventually made a full recovery from NDI with sodium level at 142mmol/L on day 27 without desmopressin (Figure 1). This case suggests that anidulafungin may be associated with partial NDI.
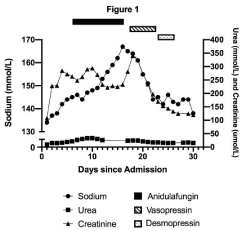
Figure 1: Plasma sodium, urea, and creatinine levels before, during, and
after anidulafungin. Anidulafungin was initiated on day 6 and discontinued
on day 16 of admission. Vasopressin infusion was started on day 17 of
admission and discontinued on day 22. Desmopressin was administered
starting day 23 and discontinued on day 26 of admission.
Discussion
Anidulafungin belongs to the echinocandin class, which includes caspofungin and micofungin. Echinocandins have favourable activity against Candida and Aspergillus species [16] and inhibit fungal cell wall formation by disrupting synthesis of beta-1,3 glucan. The halflife for elimination is forty to fifty hours. It is metabolized by a slow chemical hydrolysis into a peptide that is excreted through feces. Less than 1% of anidulafungin is excreted in urine [17].
There were no other reports of NDI associated with anidulafungin after searching databases Google Scholar, PubMed, Web of Science, and OVID. There was one case of NDI in response to Caspofungin reported to the FDA; however, the mechanism of action was not reported [18].
The proposed mechanism by which anidulafungin may cause NDI is unclear. Lithium enters the principal cells of the collecting duct and can accumulate in cytotoxic concentrations, resulting in dysregulation of aquaporin-2 water channels and impairing reabsorption of water [4]. Specifically, lithium impairs cAMP-protein kinase A mediated expression and recruitment of the aquaporin-2 channels to the apical membrane of collecting ducts [4]. Preclinical studies in rats suggest that the drug decreases cAMP formation in response to endogenous vasopressin, resulting in lower permeability of aquaporin-2 channels [20,21]. Lithium also increases expression of cyclooxygenase-2, which increases urinary prostaglandin excretion. Mice studies have shown that increased prostaglandins cause lysosomal degradation of aquaporin-2 channels [19].
The mechanism of amphotericin B causing NDI is also related to abnormal expression of aquaporin-2 channels. It could be hypothesized that the mechanism for anidulafungin-associated partial-NDI is related to down-regulation of cAMP formation leading to decreased function or reduced expression of aquaporin-2 channels. This association will need to be examined in future experimental studies, particularly because only a small percentage of anidulafungin is excreted in urine.
The diagnosis of diabetes insipidus involves a water deprivation test to differentiate DI from primary polydipsia. However, when a patient presents with hypernatremia, the water deprivation test is not necessary. In order to differentiate central versus NDI, a desmopressin challenge is used. A rise in urine osmolality of more than 100% is suggestive of central DI whereas no rise in urine osmolality indicates complete NDI. A small elevation in urine osmolality of up to 45% is suggestive of partial NDI [22]. The diagnosis of diabetes insipidus was made given the hypernatremia with high serum osmolality and relatively low urine osmolality. We reached the diagnosis of partial NDI due to the patient’s inability to maximally concentrate urine despite desmopressin infusion.
We first excluded other causes of the patient’s hypernatremia such as hypovolemia and osmotic diuresis due to the low urine osmolality and stable serum urea. Calcium and potassium levels remained in the normal range. There were no hypertonic solutions administered in the ICU. After a literature review, it was concluded that none of the patient’s other medications were known to be associated with NDI.
Renal failure causes NDI with fewer functioning nephrons leading to increased solute excretion and higher osmotic diuresis per nephron [11]. The acute kidney insult occurred on day 2 of admission and remained stable. There was no rise in creatinine alongside a rise in sodium levels. The creatinine gradually returned to baseline on day 30 of admission, after sodium had already normalized and desmopressin discontinued. Thus, we concluded that despite the initial acute kidney injury, the NDI was not related to renal failure.
The clinical suspicion that the NDI was related to anidulafungin arose due to the timing of administration of anidulafungin and the onset of hypernatremia. The delayed resolution of NDI fits with the known fifty-hour half-life of anidulafungin.
Conclusion
This case describes a patient with hypernatremia due to partial NDI following treatment with anidulafungin. Discontinuation of the offending drug and vasopressin infusion at maximal doses with adequate replacement of free water resulted in gradual correction of the hypernatremia. Timing of hypernatremia in relation to initiation of a new medication should prompt clinicians to consider medications as a potential etiology of diabetes insipidus.
References
- Sterns RH. Disorders of plasma sodium–causes, consequences, and correction. N Engl J Med. 2015; 372: 55–65.
- Bockenhauer D, Bichet DG. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol. 2015; 11: 576-588.
- Quigley J, Shelton C, Issa B. Diabetes insipidus in pregnancy. T Obstet Gyne. 2018; 20: 41-48.
- Grunfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009; 5: 270-276.
- Boton R, Gaviria M, Batlle DC. Prevalence, pathogenesis,and treatment of renal dysfunction associated with chronic lithium therapy. Am J Kidney Dis. 1987; 10: 329-345.
- Navarro JF, Quereda C, Quereda C, Gallego N, Antela A, Mora C, et al. Nephrogenic diabetes insipidus and renal tubular acidosis secondary to foscarnet therapy. Am J Kidney Dis. 1996; 27: 431-434.
- Schliefer K, Rockstroh JK, Spengler U, Sauerbruch T. Nephrogenic diabetes insipidus in a patient taking cidofovir. Lancet. 1997; 350: 413-414.
- Metzger NL, Varney Gill KL. Nephrogenic diabetes insipidus induced by two amphotericin B liposomal formulations. Pharmacotherapy. 2009; 29: 613- 620.
- Garofeanu CG, Weir M, Rosas-Arellano MP, Henson G, Garg AX, Clark WF. Causes of reversible nephrogenic diabetes insipidus: a systematic review. Am J Kidney Dis. 2005; 45: 626-637.
- Rosen S, Greenfeld Z, Bernheim J, Rathaus M, Podjarny E, Brezis M. Hypercalcemic nephropathy: chronic disease with predominant medullary inner stripe injury. Kidney Int. 1990; 37: 1067-1075.
- Tannen RL, Regal EM, Dunn MJ, Schrier RW. Vasopressin-Resistant Hyposthenuria in Advanced Chronic Renal Disease. N Engl J Med. 1969; 280: 1135-1141.
- Gabow PA, Kaehny WD, Johnson AM, Duley IT, Manco-Johnson M, Lezotte DC, et al. The clinical utility of renal concentrating capacity in polycystic kidney disease. Kidney Int. 1989; 35: 675-680.
- Scolari F, Caridi G, Rampoldi L, Tardanico R, Izzi C, Pirulli D, et al. Uromodulin storage diseases: clinical aspects and mechanisms. Am J Kidney Dis. 2004; 44: 987-999.
- Carone FA, Epstein FH. Nephrogenic diabetes insipidus caused by amyloid disease. Evidence in man of the role of the collecting ducts in concentrating urine. Am J Med. 1960; 29: 539-544.
- Frøkiaer J1, Marples D, Knepper MA, Nielsen S. Bilateral ureteral obstruction downregulates expression of vasopressin-sensitive AQP-2 water channel in rat kidney. Am J Physiol. 1996; 270: 657-668.
- Emri T, Majoros L, Toth V, Pócsi I. Echinocandins: production and applications. Appl Microbiol Biotechnol. 2013; 97: 3267-3284.
- Damle BD, Dowell JA, Walsky RL, Weber GL, Stogniew M, Inskeep PB. In vitro and in vivo studies to characterize the clearance mechanism and potential cytochrome P450 interactions of Anidulafungin. Antimicrob Agents Chemother. 2009; 53: 1149-1156.
- Commissioner, Office Of the. “Pediatrics - Safety Report Updates.” U S Food and Drug Administration. Home Page. 2018.
- Kortenoeven ML, Schweer H, Cox R, Wetzels JF, Deen PM. Lithium reduces aquaporin-2 transcription independent of prostaglandins. Am J Physiol Cell Physiol. 2012; 302: C131-140.
- Kim SW, Yeum CH, Kim S, Oh Y, Choi KC, Lee J. Amphotericin B decreases adenylyl cyclase activity and aquaporin-2 expression in rat kidney. J Lab Clin Med 2001; 138: 243-249.
- Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012; 27: 2183-2204.
- Miller M, Dalakos T, Moses AM, Fellerman H, Streeten DH. Recognition of partial defects in antidiuretic hormone secretion. Ann Intern Med. 1970; 73: 721-729.
- Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986; 108: 305-311.
- Earley LE, Orloff J. The mechanism of antidiuresis associated with the adminstration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962; 41: 1988-1997.