
Case Report
Austin J Endocrinol Diabetes. 2022; 9(1): 1094.
A Case of Pseudohypoparathyroidism - Rarely Encountered in Clinical Practice
Gomes RR*
Associate Professor, Department of Medicine, Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh
*Corresponding author: Richmond Ronald Gomes, Associate Professor, Department of Medicine, Ad-din Women’s Medical College Hospital, Dhaka, Bangladesh
Received: March 14, 2022; Accepted: April 05, 2022; Published: April 12, 2022
Abstract
Physiological intracranial calcification occurs in about 0.3-1.5% of cases. It is asymptomatic and detected incidentally by neuroimaging. Pathological basal ganglia calcification is due to various causes, such as: metabolic disorders, infectious and genetic diseases. Hypoparathyroidism and pseudohypoparathyroidism are the one of the causes of pathological basal ganglia calcification. Besides tetany and seizures this condition is presented by parkinsonism and dementia. Infections (toxoplasmosis, rubella, cytomegalovirus, cysticercosis, AIDS) give multiple and asymmetric intracranial calcification. Inherited and neurodegenerative diseases cause symmetrical, bilateral basal ganglia calcification which is not related to metabolic disorders. Fahr’s syndrome is a rare entity characterized by the presence of bilateral intracranial calcifications with predilection for the basal ganglia and dentate nuclei. It is commonly associated with endocrine disorders, particularly parathyroid and Vitamin D disturbances. Herein we report a case of pseudohypoparathyroidism revealed by Fahr’s disease.
Keywords: Basal ganglia; Calcification; Fahr’s syndrome; Pseudohypoparathyroidism (PHP)
Introduction
Fahr’s syndrome is a rare, degenerative, and neuropsychiatric disorder characterized by seizures, extrapyramidal, and neuropsychiatric symptoms as a result of symmetric and bilateral calcifications of the nucleus pallidus, the putamen, the dentate nucleus of the cerebellum (striato-pallido-dentate calcinosis) and the hemispheric white matter at the base of the skull [1,2]. Fahr’s syndrome was first reported in 1930 by Karl Theodor Fahr [3]. Diagnostic criteria of Fahr’s syndrome are as follows: bilateral calcification of the basal ganglia on neuroimaging, progressive neurologic dysfunction, absence of biochemical abnormalities, and family history consistent with autosomal dominant inheritance [4,5]. The term Fahr’s disease is used when primary familial brain calcification is present, and the term Fahr’s syndrome is used for secondary causes [6]. It is an inherited or sporadic neurological disorder with a prevalence of <1/10000002. This syndrome is mostly associated with a disorder of calcium and phosphate metabolism, especially to hypoparathyroidism and pseudohypoparathyroidism [2,7-9], but can also be attributed to other different etiologies, including infectious, metabolic, and genetic diseases [1]. PHP is a group of heterogeneous disorders with end-organ resistance of various hormones, especially parathyroid hormone (PTH) [10]. The PTH resistance usually results as hypocalcemia and hyperphosphatemia, leading to basal ganglia (BG) calcification [11]. Seizures and epilepsy occur commonly in PHP, while parkinsonism has been seldomly reported [12].
Case Presentation
An 18-year-old unmarried girl with a history of repeated hypocalcaemic seizure disorder, diagnosed at 7 years, refractory to treatment with phenytoin and valproate, presented with two new episodes of generalized tonicclonic seizures and tingling sensation over perioral regions and both upper and lower limbs for last 2 days. She did not have a history of head trauma, fever, headache, stroke, hypertension, diabetes, thyroid disease, or autoimmune disease. She did not have a family history of epilepsy; furthermore, her brothers, sisters and parents were healthy. Her birth history was uneventful with no history of any perinatal asphyxia, pre term labour or low birth weight. The patient denied any thyroid or other neck surgery. Physical examinations revealed that she had short stature (height: 152cm, weight: 62kg, body mass index (BMI): 26.8) but roundshaped face, and brachydactyly were absent. There was no knuckle dimple sign (archibald’s sign). Her vital signs were as follows: blood pressure was 110/70 mmHg; pulse rate was 74 beat per minutes; respiratory rate was 20 breaths per minutes; and body temperature was 36.3°C. Neurological examination revealed clear consciousness, with no cranial nerve abnormalities, no tremor or no bradykinesia. In addition to some cognitive impairment, on examination she had a positive Trousseau sign (flexion of the wrist and metacarpophalangeal joints when blood pressure cuff if inflated above the systolic blood pressure). Chvostek sign was absent. Laboratory studies: CBS was normal in all cell line, s. electrolyte- sodium 139mmol/l, potassium 4.4mmol/l, HCO3- 28mmol/l, s. creatinine 0.47mg/dl (normal 0.2- 1.1 mg/dl), s. calcium 6.7mg/dL (8.5-10.5 mg/dL), s. albumin 47.63 (so corrected calcium 5.7mg/dl) s. phosphorus 7.69mg/dL (2.6-4.5 mg/dL), s. magnesium 0.7mmol/l (normal 0.66-1.2 mmol/l) intact parathyroid hormone 118.6pg/dL (11-67 pg/dL) with TSH 0.607 (normal 0.35-5.5) and 25-hydroxyl vitamin D 28.33ng/dL (30-65 ng/dL). MT was non conclusive, ANA was negative (5.5U/ml). TORCH panel revealed positive IgM and IgG against HSV 1. Urinary calcium revealed hypocalciuria <2. CT of the brain revealed bilateral symmetrical calcification of the caudate nucleus, lenticular nucleus (putamen + globuspallidus), thalamus, paraventricular region (Figure 1) and also over both fronto parietal region (Figure 2). EEG revealed featured suggestive of secondarily generalized epilepsy.
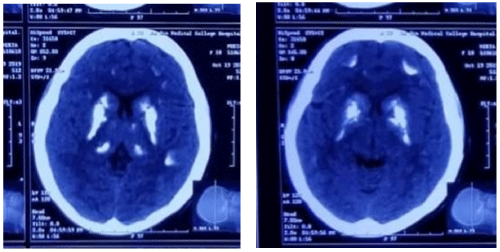
Figure 1 and 2: Showing bilateral symmetrical calcification of the caudate nucleus, lenticular nucleus (putamen + globuspallidus), thalamus, paraventricular region
and also over both fronto parietal region.
She was diagnosed with striatopallidodentatecalcinosis (or Fahr’s syndrome) secondary to hypocalcaemia due to pseudohypoparathyroidism (PHP). As the patient was having no phenotypic abnormalities so type 1 PHP (Albright’s hereditary osteodystrophy) was excluded. Differentiation between type 1b, type 1c and type 2 could not be done as there were no facilities for Ellsworth Howard test (urinary excretion of cAMP in response to intravenous PTH) and test to detect GNAS gene mutation. Patient was treated with high dose of calcium carbonate, calcitriol, phosphate binder sevelamer and anti epilepticlamotrigine. During follow up her corrected s. calcium was 9.2mg/dl, s. phosphate was 3.6mg/dl [3].
Discussion
Fahr’s syndrome is suspected to be a calcification of the basal ganglia of the brain based on brain imaging studies of patients with neurological or psychiatric symptoms such as seizure, dyskinesia, dementia, and depression. Related diseases include hyperparathyroidism, hypoparathyroidism, pseudohypoparathyroidism, hypoparathyroidism, hypervitaminosis D, tuberculosis, cytomegalovirus infection, toxoplasmosis, and astrocytoma [24,25].
Pseudohypoparathyroidim is associated with a broad spectrum of clinical manifestations. One of these is Fahr’s syndrome, that is characterized by seizures, extrapyramidal, and neuropsychiatric signs as a result of symmetric and bilateral calcifications of the nucleus pallidus, the putamen, the dentate nucleus of the cerebellum (striatopallido- dentate calcinosis), and hemispheric white matter [1,2]. In 1930, Karl Theodor Fahr, a German pathologist, first described this syndrome in a case of a man with seizures, and diffuse calcifications of brain vessels, and basal ganglia [3].
The prevalence of Fahr’s syndrome is unknown, but an incidence of basal ganglia calcifications (BGC) ranging from 0.3% to 1.2% has been reported in routine radiological examinations [13,14]. Kazis [13] analyzing 7040 computed tomography scan of the brain reported an incidence of 1.02% of symmetrical intracranial calcifications. In this study, all patients who had more extensive calcifications showed clear findings of pseudohypoparathyroidism.
The pathogenic mechanisms of BGC are unknown, although it is believed to be due to poor calcium and phosphorus control in the HPT, associated to hypokalemia, and low calcium/phosphorus ratio [7]. Active role of PTH in basal ganglia physiology may also be involved [7]. On histopathological study, Fahr’s syndrome can be labeled as intracerebral small vessel and perivascular space mineralization primarily of basal ganglia, cerebellar structures and white brain matter [15], that consists mainly of hydroxyapatite of a nature similar to that found in bones, and other elements (such as, zinc, iron, and magnesium) enmeshed in a protein rich stroma without collagen or mucopolysaccharides [16,17].
Intracranial calcifications are a common finding in neuroimaging. Although the most common cause of BGC are disorders of calcium and phosphate metabolism, more than fifty clinical conditions have been associated with BGC, including inflammatory, infectious, tumoral, endocrine, hypoxic, vascular, toxic, metabolic, and genetic disorders [1,18]. Among these conditions, the endocrine diseases, hypoparathyroidism, hyperparathyroidism and pseudohypoparathyroidism are the most common causes of Fahr’s syndrome [2,7-9].
About half of the patients with BGC exhibit neuropsychiatric manifestations and deficits. The most common neurological symptoms are headache, vertigo, movement disorders (such as Parkinson’s tremor, dystonia, and cerebellar ataxia), myoclonus and seizures [1]. Other neurological symptoms consist of stroke like events, speech disorders, coma, cognitive impairment, and syncope [18-21]. The psychiatric symptoms are usually more prominent than the neurologic symptoms in the onset of the disease [8,9]. Forty percent of patients with BGC present themselves with psychiatric symptoms at the beginning of the disease, and usually precede neurological manifestations [8,9]. The psychiatric symptoms may be composed of a wide variety of symptoms ranging from depression to psychosis [2,8,9,21,22]. Among these, cognitive and psychotic disorders are most prominent [8,9].
In the presence of BGC, especially with symptoms, laboratory investigation should be performed to assess the occurrence of hypoparathyroidism and pseudohypoparathyroidism. The typical findings in untreated pseudohypoparathyroidm are high intact PTH levels, hypocalcemia, hyperphosphatemia, normal levels of 1, 25-dihydroxyvitamin D, and relatively low urinary calcium excretion. Laboratory evaluation also should include serum creatinine, and magnesium levels.
The treatment of Fahr’s syndrome is directed to the identifiable cause [23]. Especially in hypoparathyroidism and pseudohypoparathyroidism, the early treatment can prevent calcifications and neurophysiological disorders [2,3,21]. Goswami [3] reported a reduced risk of BCG progression with treatment to restore calcium/phosphorus ratio in patients with idiopathic HPT. In symptomatic patients, seizures show poor response to anticonvulsants, and resolution when normal plasma calcium level is reached [2]. Parkinsonism is also characterized by resistance to standard treatment, and in early cases may respond to calcium therapy [2]. The psychiatric symptoms usually improve with normalization of calcemia, although the dementia has the worst response [2]. It is noteworthy that the prognosis is variable, cannot be predicted and is unrelated to the extent of calcifications [21]. Death is usually due to neurological deterioration [21].
PHP is classified as different subtypes, including Ia (Albright hereditary osteodystrophy), Ib, Ic, and PHP 2 [12]. PHP Ia was the most common subtype and accompanied by a blunted cAMP and phosphaturic response to exogenous PTH due to the end-organ resistance. Besides, the patient of PHP Ia usually had severe albrights hereditary osteodystrophy (AHO), presenting with heterogeneous clinical manifestations, such as short stature, brachydactyly, and soft tissue calcification [10]. Genetically, PHP Ia was usually caused by genetic alterations within or upstream of the imprinted GNAS gene, which encodes the G-stimulatory protein alpha (Gs-a) [26]. It is also associated with thyroid stimulating hormone resistance. Type 1b Lacks the physical appearance of type 1a, but is biochemically similar. It is associated with a methylation defect in the A/B exon of GNAS1, caused by STX16 disruption. Type 2 also lacks the physical appearance of type 1a. Since the genetic defect in type 2 is further down the signaling pathway than in type 1, there is a normal cAMP response to PTH stimulation despite the inherent abnormality in calcium regulation. The specific gene is not identified. While biochemically similar, type 1 and 2 disease may be distinguished by the differing urinary excretion of cyclic AMP in response to exogenous PTH.
Some sources also refer to a “type 1c” [30]. The phenotype is the same as in type 1a, but red blood cells show normal Gs activity. As it is also caused by a GNAS mutation, it is not clear whether it should be considered an entity separate from Ia [31].
Brain calcification was usually investigated in pseudohypoparathyroidism due to the hyperphosphatemia. However, in most conditions, seizure and epilepsy are the common clinical manifestations resulting from the calcification of subcortical region and BG, other than parkinsonism. Few cases were ever reported in this condition. Bradley reported a case in PHP presenting as parkinsonism, without BG calcification [27], suggesting that the mechanism might also have other pathophysiology, such as impaired neurotransmission.
In addition, the calcification of BG presenting with parkinsonism can also be caused by various of other diseases, such as familial idiopathic BG calcification (Fahr disease), neoplastic, vascular, infectious, and congenital causes, as well as other endocrine/ metabolic diseases, including diabetes mellitus, hypoparathyroidism, and hyperparathyroidism [5] Apart from the bilateral BG, the diagnosis criteria of Fahr disease usually contain family history, neuropsychiatric symptoms, and no underlying systemic etiology [28]. Further evaluations should be conducted to distinguish PHP from the diseases mentioned above, including serum levels of calcium, phosphate, PTH, TSH, alkaline phosphatase, and calcitonin; CSF examinations; metabolic, inflammatory, and infectious conditions; and heavy mental concentrations [29].
Overall, PHP is a rare etiology for parkinsonism with calcification of BG. However, markedly improvement could be investigated after the correction of hypocalcemia and hyperphosphatemia. Thus, it is of great meaningful to differentiate PHP from other parkinsonism, and a family history of seizures may be an indicator for consideration, in addition, several laboratory examinations and brain CT scan can be conducted if necessary.
Conclusion
Despite its rarety, the possibility of Fahr’s syndrome should be considered in patients with neuropsychiatric disturbances, seizure disorder. In presence of symmetrical and abnormal bilateral basal ganglia calcification, hypothyroidism, hypoparathyroidism and pseudohypoparathyroism should be investigated. This case report stresses the major benefits of calcium supplementation and calcitriol, phosphate binder and anti epileptic, even with the patient being in advanced stage of disease.
References
- Swami A, Kar G. Intracranial hemorrhage revealing pseudohypoparathyroidism as a cause of fahr syndrome. Case Rep Neurol Med. 2011; 2011: 407567.
- Ramisa J, Ibanez AE, Irigoína RR, Artigasa CF, Comasa LM. Extrapyramidal symptoms due to calcinosiscerebri in a patient with unknown primary hypoparathyroidism. Endocrinol Nutr. 2012; 59: 69-71.
- Fahr T. Idiopathischeverkalkung der hirngefässe. Zentrabl Allg Pathol. 1930; 50: 129-133.
- Shafaq S, Hafiz MA, Maheen A, et al. Fahr’s syndrome: Literature review of current evidence. Orphanet J Rare Dis. 2013; 8: 156.
- Mufaddel AA, Alhassani GA. Familial idiopathic basal ganglia calcification (Fahr’s disease). Neurosciences (Riyadh). 2014; 19: 171-177.
- Perugula ML, Lippmann S. Fahr’s disease or Fahr’s syndrome? Innov Clin Neurosci. 2016; 13: 45-46.
- Goswami R, Sharma R, Sreenivas V, Gupta N, Ganapathy A, Das S. Prevalence and progression of basal ganglia calcification and its pathogenic mechanism in patients with idiopathic hypoparathyroidism. Clin Endocrinol (Oxf). 2012; 77: 200-206.
- Srivastava S, Bhatia MS, Sharma V, Mahajan S, Rajender G. Fahr’s Disease: An Incidental Finding in a Case Presenting with Psychosis. Ger J Psychiatry. 2010; 13: 86-90.
- Senoglu M, Tuncel D, Orhan FO, Yuksel Z, Gokçe M. Fahr’s Syndrome: A Report of Two Cases. Firat Tip Dergisi. 2007; 12: 70-72.
- Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab. 2011; 96: 3020-3030.
- Maeda SS, Fortes EM, Oliveira UM, et al. Hypoparathyroidism and pseudohypoparathyroidism. Arq Bras Endocrinol Metabol. 2006; 50: 664-673.
- Ritter C, Gobel CH, Liebig T, et al. An epigenetic cause of seizures and brain calcification: pseudohypoparathyroidism. Lancet. 2015; 385: 1802.
- Kazis AD. Contribution of CT scan to the diagnosis of Fahr’s syndrome. Acta Neurol Scand. 1985; 71: 206-211.
- Fenelon G, Gray F, Paillard F, Thibierge M, Mahieux F, Guillani A. A prospective study of patients with CT detected pallidal calcifications. J NeurolNeurosurg Psychiatry. 1993; 56: 622-625.
- Unkrig S, Gullotta F, Madea B. MorbusFahr-considerations on a case of sudden death. Forensic Sci Int. 2011; 204: e12-15.
- Konig P. Psychopathological alterations in cases of symmetrical basal ganglia sclerosis. Biol Psychiatry. 1989; 25: 459-468.
- Smeyers-Verbeke J, Michotte Y, Pelsmaeckers J, Lowenthal A, Massart DL, Dekegel D, et al. The chemical composition of idiopathic nonarteriosclerotic cerebral calcifications. Neurology. 1975; 25: 48-57.
- Baba Y, Broderick DF, Uitti RJ, Hutton ML, Wszolek ZK. Heredofamilial brain calcinosis syndrome. Mayo Clin Proc. 2005; 80: 641-651.
- Castro MC, Singer BH. Agricultural settlement and soil quality in the Brazilian Amazon. Popul Environ. 2012; 34: 22-43.
- Al-Jehani H, Ajlan A, Sinclair D. Fahr’s Disease Presenting with Aneurysmal Subarachnoid Hemorrhage. J Clin Imaging Sci. 2012; 2: 27.
- Rastogi R, Singh AK, Rastogi UC, Mohan C, Rastogi V. Fahr’ssyndrome: a rare clinico-radiologicentity. MJAFI. 2011; 67: 159-161.
- Gulsun M, Baykiz AF, Kabatas S, Belli H. Fahr’s Syndrome - three cases presenting with psychiatric signs. Eur J Gen Med. 2006; 3: 35-40.
- Mitchell DM, Regan S, Cooley MR, Lauter KB, Vrla MC, Becker CB, et al. Long-term follow-up of patients with hypoparathyroidism. J Clin Endocrinol Metab. 2012; 97: 4507-4514.
- Moriwaki Y, Matsui K, Yamamoto T, et al. Cerebral subcortical calcification and hypoparathyroidism-a case report and review of the literature. Jpn J Med. 1985; 24: 53-56.
- Sava A, Dumitrescu G, Haba D, et al. The Fahr syndrome and the chronic lymphocytic thyroiditis. Rom J Morphol Embryol. 2013; 54: 195-200.
- Elli FM, Linglart A, Garin I, et al. The prevalence of GNAS deficiency-related diseases in a large cohort of patients characterized by the EuroPHP network. J Clin Endocrinol Metab. 2016; 101: 3657-3668.
- Evans BK, Donley DK. Pseudohypoparathyroidism, Parkinsonism syndrome, with no basal ganglia calcification. J Neurol Neurosurg Psychiatry. 1988; 51: 709-713.
- Manyam BV. What is and what is not “Fahr’s disease”. Parkinsonism Related Disord. 2005; 11: 73-80.
- Bonazza S, La Morgia C, Martinelli P, et al. Strio-pallido-dentate calcinosis: a diagnostic approach in adult patients. Neurol Sci. 2011; 32: 537-545.
- Aldred MA. “Genetics of pseudohypoparathyroidism types Ia and Ic”. J. Pediatr. Endocrinol.Metab. 2006; 19: 635-640.
- Bastepe M. “The GNAS locus and pseudohypoparathyroidism”. Advances in Experimental Medicine and Biology. 2008; 626: 27-40.