
Review Article
J Endocr Disord. 2014;1(1): 1006.
New Mechanistic Insights in the Development of Diabetic Nephropathy: Role of Cytochromes P450 and Their Metabolites
Stephanie Eid1§, Cesar Abdul-Massih1§, Christopher M El-Khuri1§, Ahmed Hamdy2, Awad Rashid2 and Assaad A Eid1*
1Department of Anatomy Cell Biology and Physiology, American University, Lebanon
2Department of Nephrology, Hamad Medical Corporation, Qatar
3The Authors Equally Contributed to This Work.
*Corresponding author: Assaad A. Eid, Faculty of Medicine, Department of Anatomy, Cell Biology and Physiology, American University of Beirut, Bliss Street, 11-0236, Riad El-Solh 1107-2020, Lebanon
Received: Aug 08, 2014; Accepted: Aug 27, 2014; Published: Aug 28, 2014
Abstract
Diabetic Nephropathy (DN), a major complication of diabetes, is characterized by hypertrophy, extracellular matrix accumulation, fibrosis and proteinuria leading to loss of renal function. However, the mechanisms leading to kidney injury is not well defined. Arachidonic acid is primarily metabolized by Cytochromep-450 (CYP) enzymes to 20-Hydroxyeicosatetraenoic acid (20-HETE) and Epoxyeicosatrienoic acids (EETs) and these compounds have a role in renal physiology and pathophysiology. Recent studies show that the production of EETs and 20-HETE is altered in diabetes. More interestingly, we and others have established a link between the alteration in the production of 20-HETE and EETs and the onset and development of diabetic-induced kidney injury, and that these cytochromes P450 metabolites of arachidonic acid contribute to the changes in renal structure and function seen in diabetic nephropathy. All the results described in this review suggest that the drugs that modify the formation and/or actions of EETs and 20-HETE may have therapeutic benefits for diabetic nephropathy treatment.
Keywords: Diabetic Nephropathy; Reactive Oxygen Species (ROS); Cytochromes P450; 20-HETE; EETs
Diabetes-related Kidney Disease or Diabetic Nephropathy
Diabetes is a major public health problem that affects about 8% of the world population. An estimated 24.1 million people have diabetes [1]. The incidence of diabetes has increased tremendously in the past 10 years. This alone makes it an epidemic disease. Diabetes is associated with a number of metabolic risk factors that contribute to a high rate of micro and macro vascular events.
Diabetes-related kidney disease or Diabetic Nephropathy (DN) is a major risk factor for cardiovascular morbidity and mortality and is considered to be one of the most serious complications of diabetes worldwide, affecting up to 25% and 40% of all patients with type 1 and type 2 diabetes, respectively [2]. DN is characterized by increased urinary albumin excretion (microalbuminuria), which often progresses to proteinuria, one of the most important prognostic risk factors for kidney disease progression [3]. Microalbuminuria/ proteinuria develops as a consequence of a series of structural changes in the vascular, glomerular, tubular and interstitial compartments in the absence of measurable dysfunction [4].
The glomerular changes include hyperplasia/hypertrophy with thickening of glomerular basement membrane, mesangial expansion with extracellular matrix accumulation, changes in glomerular epithelial cells (podocytes), including a decrease in number and/ or density, podocyte foot process broadening and effacement and glomerulosclerosis. Similar changes occur in the tubulointerstitial compartment and include matrix protein accumulation, thickening of tubular basement membranes and interstitial fibrosis [5].
Although the mechanisms of renal cell injury in DN are not fully understood, much attention has focused on the role of high glucose per se. Tubular as well as glomerular cells are primary target of hyperglycemia and chronicexposure to elevated blood glucose levels contributes to the functional and phenotypic changes seen in overt DN.
Reactive Oxygen Species in the Development of Diabetic Nephropathy
There is increasing evidence that the overproduction of Reactive Oxygen Species (ROS) is a major factor in the development of diabetic vascular complications as well as diabetes per se [6]. It is now widely recognized that alteration in ROS production in diabetes is a direct consequence of hyperglycemia [6] and that various types of cells including glomerular cells (endothelial, mesangial, and epithelial) as well as tubular epithelial cells are capable of producing ROS in the diabetic milieu.Oxidative stress is thought to be a critical factor in the development of DN. Increased generation of reactive oxygen species occurs in the kidney concomitant with disease progression. Antioxidants prevent glomerular and renal hypertrophy, as well as proteinuria, suggesting a pathogenic role for ROS in the progression of diabetes [7-13].
In the present review, we will focus on CYP metabolites of AA as significant sources of ROS in the kidney in addition to their functional and signaling roles in renal glomerular and tubular cells in various experimental models of nephropathy, including studies by our group.
Metabolism of Arachidonic Acid by Cytochrome P450 Enzymes
Arachidonic Acid (AA), a lipid released as a byproduct of phospholipases, can be metabolized by several pathways: The cyclooxygenase, the lipoxygenase or the Cytochrome P450 (CYP) monooxygenase pathway. In the latter, AA is metabolized by several CYP450 is forms to produce 5,6-, 8,9-, 11,12- and 14,15-Epoxyeicosatrienoic acids (EETs), dihydroxyeicosatetraenoic acids, and 19- and20-HETEs [14,15] (Figure 1).
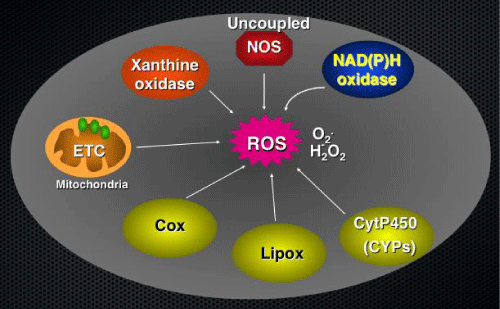
Figure 1: Potential sources of Reactive Oxygen Species (ROS) in cells. Many enzymes, including those in the mitochondrial electron transport chain, xanthine oxidase, uncoupled NOS, NAD(P)H oxidases, lipoxygenases, cyclooxygenases and cytochrome P450 monooxygenase, produce ROS in different organs of the body including the kidneys.
Cytochrome P450s (P450) have been originally described to play a role in the metabolism of toxic chemicals, drugs, and endogenous substrates, such as steroids and cholesterol [16]. Cytochromes P450s are expressed in a gender-, age-, and organ-specific manner and their levels are regulated by hormones, cytokines, diet, fasting, drugs, or are altered during the course of diseases, such as obesity, diabetes, and hypertension. However, our knowledge of the physiological and/ or pathophysiological significance of these proteins is still under investigation. The identification of EETs and 20-HETE, as products of AA metabolism in rodent and human tissues, established the AA monooxygenase as a formal metabolic pathway, and suggested a biological role for its metabolites [16]. 20-HETE is one of the main AA metabolites produced in the kidney cortex. It is synthesized by CYPs of the 4A and 4F subfamilies, which are highly expressed in the renal cortex [17-21]. EETs are mainly synthesized by CYPs of the 1A, 2B, 2C and 2J subfamilies [15,22]. Studies have shown that CYPs 2C and 2J are the predominant isoforms of cytochromes expressed in the kidney [23,24]. 20-HETE and EETs exert a wide range of regulatory and opposing functions depending on the location of their productions. Recent studies on the functional role of CYPs 450 and their metabolites provided new avenues on the role of these enzymes in the pathophysiology of certain diseases, i.e. diabetes, cancer, hypertension, etc.
Pathophysiological Role of 20-HETE and EETs in the Kidneys
The expression of CYP450 enzymes and their metabolites in the kidney is altered in many kidney diseases, i.e. diabetes, pregnancy, hepatorenal syndrome, cyclosporin-induced nephrotoxicity, polycystic kidney disease and hypertension [25-27]. These findings suggest that the alterations in the renal production of 20-HETE and EETs may contribute to the (decline/abnormalities) in kidney functions that are associated with these conditions.
20 HETE
20-HETE is a potent vasoconstrictor known to play key roles in both tubular and vascular regulation of renal hemodynamic and extracellular fluid volume [28]. 20-HETE can play opposite roles in kidney homeostasis depending on the cell type that produces and/ or targets this AA-derived lipid. Several studies have implicated20- HETE as a potential mediator of cellular proliferation in the pathogenenesis of many kidney diseases, including polycystic kidney disease [29] and renal cell carcinoma [30]. Also, CYP450 inhibitors attenuate 20-HETE-mediated renal cell proliferation.
In addition to its role in cellular proliferation, 20-HETE has been shown to exert both prohypertensive and antihypertensive actions [31-35]. The inhibition of 20-HETE synthesis significantly reduced the development of hypertension and had considerable nephroprotective and cardio protective effects, including reduction of proteinuria, glomerulosclerosis and tubulointerstitial injury [36]. In contrast, another study showed that up regulation of TGF-β in Dahl salt-sensitive rats may induce the development of proteinuria and renal disease in part by inhibiting the formation of 20-HETE [37].
In addition to the renal tubules and vasculature, glomeruli also produce 20-HETE, which can either protect the kidney from injury-mediated albuminuria or contribute to injury by promoting high glucose–mediated podocyte apoptosis or tubular hypertrophy [7,9,10].
EETs
EETs synthesized by the vascular endothelium are considered potent vasodilators [38-40]. In parallel to their hemodynamic role, EETs have been reported to exert diverse biological effects in the renal system, such as anti-inflammatory, anti-apoptotic, pro-proliferative, pro-angiogenic, anti-oxidative, anti-fibrotic, and anti-hypertensive effects [41-46].Therefore, like 20-HETE, aberrant levels of EETs might be expected to alter cellular function.
Substantial evidence indicates that EETs have renoprotective potential. The pharmacological inhibition of soluble Epoxide Hydrolase (sEH), an enzyme that metabolizes EETs to their less biologically active dihydroxyeicosatrienoic acid metabolite, increases EET bioavailability and provides anti-inflammatory, antioxidative, and antiapoptotic activities in a number of kidney diseases. Cisplatininduced renal injury is reduced upon the administration of sEH inhibitors [47,48].
Another study showed that the inhibition of sEH in hypertensive Goto-Kakizaki rats attenuated the progression of glomerular and tubular injury associated with hypertension and diabetes [49]. Also, the over expression of CYP2J2, which increases EET production, protects against renal damage in a rat 5/6 nephrectomy (5/6-Nx) model of chronic renal failure by inhibiting renal cell apoptosis and tubule interstitial fibrosis [44].
20-HETE, EETs and Reactive Oxygen Species
There are multiple sources of ROS in cells and tissues (Figure 2). In addition to the mitochondria, the peroxisomes, and the NADPH oxidizes family, Cytochromes P450 have been shown to be significant sources of oxidative stress in kidneys, liver and coronary arteries [7,50,51]. Being home-containing monooxygenases, CYPs activate oxygen prior to incorporating it into a substrate; futile or redox cycling of the enzyme, where activated oxygen “escapes” before incorporation into organic products, leads to the formation of superoxide/H2O2(Figure 3).

Figure 2: Metabolism of Arachidonic Acid by the CYP mono-oxygenases. CYP-450 epoxygenases, CYP1A, 2B, C and J, produce EETs while the ω-hydroxylases, CYP4A and F, produce 19 and 20-HETE.
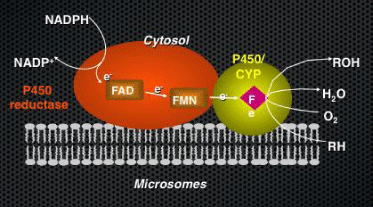
Figure 3: CYP mono-oxygenases as a source of Reactive Oxygen Species (ROS). CYP genes encode for membrane-bound, heme-containing enzymes and require NADPH reductase and CYPb5 (NADH/NADPH oxidase) as cofactors. The binding of the substrate (RH) to ferric P450 results in the formation of the substrate complex. The ferric P450 then accepts the first electron from cytochrome P450 reductase and is reduced to the ferrous intermediate. This intermediate then binds an oxygen molecule to form oxycomplex, which is further reduced to give peroxycomplex. The input of protons to this intermediate can result in the heterolytic cleavage of the O–O bond, producing H2O and the ‘oxenoid’ complex, the latter of which theninserts the heme-bound activated oxygen atom into the substrate molecule to produce ROH [68].
Under the pathological condition of renal ischemia, 20-HETE mediated the production of ROS and induced cytotoxicity and apoptosis of tubular epithelial cells [52,53]. The changes seen in the kidney have been also demonstrated on heart cells, in cardiac studies. The oxidative stress mediated by 20-HETE was shown to play a role in myocardial injury during ischemia-reperfusion in myocytes as well [54].
The involvement of CYPs family and their metabolites in glomerular epithelial cells as well as in tubular epithelial cells to produce significant ROS was recently described by our group [7,9,10]. In glomerular epithelial cells as well as in tubular epithelial cells, our group showed that 20-HETE increases ROS production [7,9,10]. In these studies, Eid AA et al. and Eid S. et al show that 20-HETE exerts is deleterious effects on glomerular epithelial cells as well as tubular epithelial cells through the generation of reactive oxygen species (Figure 4).
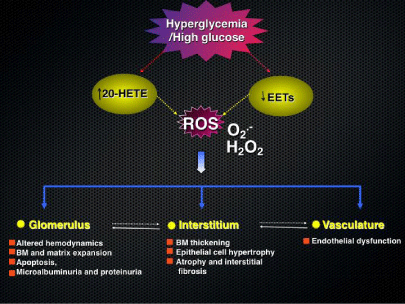
Figure 4: Proposed mechanisms of diabetes induced renal Injury. Role of cytochromes p450 enzymes and their metabolites in diabetes-induced renal injury [7,9,10].
Little is known about the role of EETs and its correlation with oxidative stress. EETs have been shown to inhibit apoptosis and to reduce the levels of oxidative stress in the long after ischemia/ reperfusion [55]. Also, Epoxyeicosatrienoic Acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide [56]. In the kidney, Eid S et al. established that the inhibition of EETs induces ROS production and mediates the deleterious effect of HG on proximal tubular cells. Blockade of EETs in a HG milieu is associated with increased ROS generation, which may account for the resulting proximal tubular cell injury in the hyperglycemic milieu [10].
Alternatively, these CYPs or the products that they form might also be sensitive to the presence of ROS produced by other sources, especially the nitric oxide syntheses leading to the exacerbation of disease symptoms. Indeed, superoxide has been shown to inhibit 20- HETE productions by renal cortical macrodomes [57], as well as to enhance breakdown of 20-HETE to a more polar product, such as the 20-carboxylic acid. Thus, lipid per oxidation of CYP-generated HETEs and EETs by ROS might contribute to the overall decline in renal function.
Role of 20-HETE and EETs in Diabetic Nephropathy
Recent studies have shown a correlation between diabetic nephropathy and altered expression and activity in CYP450s and their metabolites 20-HETE and EETs [9,10]. Pathological changes and alterations in both glomerular and tubular compartments have been demonstrated to be linked to these metabolites.
Several research groups, including ours, have implicated alterations in CYPs450 metabolites as contributing to renal damage in diabetes. Enriquez et al. described elevated CYP4A expression accompanied by an increase in 20-HETE generation in renal microsomes in streptozotocin induced diabetic rats [58,59]. However the role of 20-HETE in diabetic kidney disease is still controversial. In some studies, 20-HETE has been described to play a major role in the development of proteinuria, hallmark of kidney disease progression especially in diabetes. Ex vivo experiments showed that 20-HETE reduces albumin permeability in isolated glomeruli from Puromycin Amino Nucleoside (PAN)-induced nephritic syndrome in animal models [60]. In parallel with these findings, other investigators showed that the development of proteinuria and glomerular injury resulted from the inhibition of the glomerular production of 20- HETE by increased TGF-β1[37]. In contrast, our group demonstrated that high glucose induces ROS production and apoptosis in cultured mouse podocytes through the up regulation of CYP4A with increased production of 20-HETE. Inhibition of CYP4A prevented oxidative stress and podocyte apoptosis in vitro and reduced albumin excretion and podocyte injury and depletion in OVE26 mice, a well-established model of type 1 diabetes [7]. Taken together these data suggest that altered 20-HETE levels may be a general characteristic of protein uric states.
Besides glomerular injury, proximal tubular epithelial cell hypertrophy and eventual atrophy as well as apoptosis are features of DN. It has been documented that 20-HETE, by generating ROS, mediates the cytotoxicity and apoptosis of tubular epithelial cells seen during renal ischemia [52,53]. In the context of diabetic nephropathy, diabetes up regulates CYP4A expression in the kidney, leading to increased production of 20-HETE. Inhibition of CYP4A ameliorates tubular renal hypertrophy, and prevents oxidative stress by inhibiting NADPH oxidase activation and the increase in TGF-β1 expression and levels, suggesting that diabetes induces kidney hypertrophy and tubulointerstitial changes through the generation of ROS via up regulation of CYP4A and NADPH oxidase. The authors [9] suggest that up regulation of CYP4A expression and increase in 20-HETE formation leading to increased ROS production coincide with increased TGF-β expression, and that these changes were prevented by inhibition of CYP4A. In the kidney, TGF-β1 promotes hypertrophy and regulates production of collagen and fibronectin, important contributors to the causation of the kidney damage seen in DN [61]. The role of 20-HETE in regulating TGF-β1 is controversial. In contrast to the finding where Eid S et al. showed that hyperglycemia-induced kidney hypertrophy and tubular injury is mediated by increased CYP4A/20-HETE formation, which in turn regulate the deleterious effect of TGF-β1 in the development of diabetic nephropathy, Dahly-Vernon et al [37] showed that TGF-β1 contributes to the development of glomerular injury in Dahl saltsensitive rats by increasing glomerular permeability to albumin through the inhibition of glomerular 20-HETE formations. In another study, the authors show that 20-HETE inhibits the proliferation of vascular smooth muscle cells via transforming growth factor-β [62].
The precise role of EETs in the progression of DN is still not well characterized. The inhibition of the enzyme sEH, causing a secondary increase in EETs production, induced albuminuria in mice models of progressive renal disease [63]. In another study, Williams et al. [64] showed that EETs play an essential role in the maintenance of the glomerular permeability barrier to albumin. In a comparable study, Luo et al. [65] noted that a decrease in EETs production is coupled with an increase in TGF-β production and alters the permeability of the glomerular barrier and adds to the glomerular damage seen early on in diabetic nephropathy. In contrast, recent data show that the over expression of CYP2J2 epoxygenase followed by increased EET generation in streptozotocin-induced diabetic mice attenuated microalbuminuria and glomerulosclerosis progression via inhibition of TGF-β/Smad signaling [66]. In the same line of work, our group demonstrated that tubulointerstitial changes and tubular hypertrophy are associated with a significant alteration in the expression and activity of cytochromes P450 of the 2C family and EETs production, an effect mediated through the generation of ROS alteration. The authors show that chronic exposure of renal epithelial cells to HG reduces CYP2C11 protein expression and EETs formation. This was paralleled by stimulation of ROS production, increase in extracellular matrix protein expression, and cellular hypertrophy. More interestingly, the use of MSPPOH, a competitive inhibitor of epoxygenase activity, in the hyperglycemic milieu, induces tubulointerstitial changes and increases tubular injury. Also, it has been shown that in humans with renovascular disease, circulating levels of 20-HETE are increased and those of EETs are decreased; urinary excretion of 20-HETE is also decreased suggesting that alteration in 20-HETE and EETs may contribute to the vascular and tubular abnormalities of renovascular disease [67].
Taken together, and despite the controversial findings, all these studies feature the important role played by Cytochromes P450 enzymes and their AA metabolites in the onset and development of diabetic nephropathy.
Conclusion
During the onset and development of diabetes, injury of kidney cells leads to kidneys dysfunction and end stages renal disease. While the control of blood sugar in diabetes is essential, strict control is difficult to achieve and blockade of the renin/angiotensin system does not result in complete protection. Therefore, the results highlighted in this review, suggest that understanding the alteration in cytochromes P450 enzymes and their metabolites during the onset and development of DN may be useful in designing effective therapies, in addition to the metabolic control drugs, in the treatment of DN.
Acknowledgment
The authors are supported by: Regular research grant from Qatar National Research Foundation (NPRP# 5-410-3-113) for AAE and AR, and a regular research grant from the National Center for Scientific Research Lebanon for AAE.
References
- International Diabetes Federation. IDF Diabetes Atlas, 6th edn. Brussels, Belgium: International Diabetes Federation. 2013.
- Hall P. Prevention of progression in diabetic nephropathy. Diabetes Spectrum. 2006; 19: 18-24.
- de Zeeuw D. Albuminuria, not only a cardiovascular/renal risk marker, but also a target for treatment? Kidney Int Suppl. 2004; 2-6.
- Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999; 56: 1627-1637.
- Jones SC, Saunders HJ, Pollock CA. High glucose increases growth and collagen synthesis in cultured human tubulointerstitial cells. Diabet Med. 1999; 16: 932-938.
- Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a congress series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diab./Metab. Res. Rev. 2001; 17: 189–212.
- Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, et al. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009; 58: 1201-1211.
- Eid AA, Ford BM, Block K, Kasinath BS, Gorin Y, Ghosh-Choudhury G, et al. AMP-activated protein kinase (AMPK) negatively regulates Nox4-dependent activation of p53 and epithelial cell apoptosis in diabetes. Journal of Biological Chemistry. 2010; 285: 37503-37512.
- Eid S, Abou-Kheir W, Sabra R, Daoud G, Jaffa A, Ziyadeh FN, et al. Involvement of renal cytochromes P450 and arachidonic acid metabolites in diabetic nephropathy. J Biol Regul Homeost Agents. 2013; 27: 693-703.
- Eid S, Maalouf R, Jaffa AA, Nassif J, Hamdy A, Rashid A, et al. 20-HETE and EETs in diabetic nephropathy: a novel mechanistic pathway. PLoS One. 2013; 8: 70029.
- Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, et al. Nox4 NAD (P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem. 2005; 280: 39616-39626.
- Gorin Y, Block K. Nox4 and diabetic nephropathy: With a friend like this, who needs enemies? Free Radic Biol Med. 2013; 61: 130-142.
- Eid AA, Ford BM, Bhandary B, de Cassia Cavaglieri R, Block K, Barnes JL, et al. Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes. 2013; 62: 2935-2947.
- Zeldin DC. Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem. 2001; 276: 36059-36062.
- Natarajan R, Reddy MA. HETEs/EETs in renal glomerular and epithelial cell functions. Curr Opin Pharmacol. 2003; 3: 198-203.
- Capdevila JH, Falck JR, Imig JD. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007; 72: 683-689.
- Powell PK, Wolf I, Lasker JM. Identification of CYP4A11 as the major lauric acid omega-hydroxylase in human liver microsomes. Arch Biochem Biophys. 1996; 335: 219-226.
- Schwartzman ML, da Silva JL, Lin F, Nishimura M, Abraham NG. Cytochrome P450 4A expression and arachidonic acid omega-hydroxylation in the kidney of the spontaneously hypertensive rat. Nephron. 1996; 73: 652-663.
- Christmas P, Jones JP, Patten CJ, Rock DA, Zheng Y, Cheng SM, et al. Alternative splicing determines the function of CYP4F3 by switching substrate specificity. Journal of Biological Chemistry. 2001; 276: 38166-38172.
- Kalsotra A, Cui X, Anakk S, Hinojos CA, Doris PA, Strobel HW. Renal localization, expression, and developmental regulation of P450 4F cytochromes in three strains of spontaneous hypertensive rats. Biochemical and Biophysical Research Communications. 2005; 338, 423-431.
- Ito O, Nakamura Y, Tan L, Ishizuka T, Sasaki Y, Minami N, et al. Expression of cytochrome P-450 4 enzymes in the kidney and liver: regulation by PPAR and species-difference between rat and human. Mol Cell Biochem. 2006; 284: 141-148.
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002; 82: 131-185.
- Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004; 52: 447-454.
- Imaoka S, Hashizume T, Funae Y. Localization of rat cytochrome P450 in various tissues and comparison of arachidonic acid metabolism by rat P450 with that by human P450 orthologs. Drug Metab Pharmacokinet. 2005; 20: 478-484.
- Roman RJ, Alonso-Galicia M. P-450 Eicosanoids: A Novel Signaling Pathway Regulating Renal Function. News Physiol Sci. 1999; 14: 238-242.
- McGiff JC, Quilley J. 20-HETE and the kidney: resolution of old problems and new beginnings. Am J Physiol. 1999; 277: 607-623.
- Maier KG, Roman RJ. Cytochrome P450 metabolites of arachidonic acid in the control of renal function. Curr Opin Nephrol Hypertens. 2001; 10: 81-87.
- Carroll MA1, McGiff JC. A new class of lipid mediators: cytochrome P450 arachidonate metabolites. Thorax. 2000; 55: 13-16.
- Park F, Sweeney WE, Jia G, Roman RJ, Avner ED. 20-HETE mediates proliferation of renal epithelial cells in polycystic kidney disease. J Am Soc Nephrol. 2008; 19: 1929-1939.
- Alexanian A, Rufanova VA, Miller B, Flasch A, Roman RJ, Sorokin A. Down-regulation of 20-HETE synthesis and signaling inhibits renal adenocarcinoma cell proliferation and tumor growth. Anticancer Res. 2009; 29: 3819-3824.
- Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML. 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev. 2014; 22: 1-12.
- Ward NC, Rivera J, Hodgson J, Puddey IB, Beilin LJ, Falck JR, et al. Urinary 20-hydroxyeicosatetraenoic acid is associated with endothelial dysfunction in humans. Circulation. 2004; 110: 438-443.
- Nowicki S, Chen SL, Aizman O, Cheng XJ, Li D, Nowicki C, et al. 20-Hydroxyeicosa-tetraenoic acid (20 HETE) activates protein kinase C. Role in regulation of rat renal Na+,K+-ATPase. J Clin Invest. 1997; 99: 1224-1230.
- Yu M, Lopez B, Dos Santos EA, Falck JR, Roman RJ. Effects of 20-HETE on Na+ transport and Na+ -K+ -ATPase activity in the thick ascending loop of Henle. Am J Physiol Regul Integr Comp Physiol. 2007; 292: 2400-2405.
- Ominato M, Satoh T, Katz AI. Regulation of Na-K-ATPase activity in the proximal tubule: role of the protein kinase C pathway and of eicosanoids. J Membr Biol. 1996; 152: 235-243.
- Certíková Chábová V, Walkowska A, Kompanowska-Jezierska E, Sadowski J, Kujal P, Vernerová Z, et al. Combined inhibition of 20-hydroxyeicosatetraenoic acid formation and of epoxyeicosatrienoic acids degradation attenuates hypertension and hypertension-induced end-organ damage in Ren-2 transgenic rats. Clin Sci (Lond). 2010; 118: 617-632.
- Dahly-Vernon AJ, Sharma M, McCarthy ET, Savin VJ, Ledbetter SR, Roman RJ. Transforming growth factor-beta, 20-HETE interaction, and glomerular injury in Dahl salt-sensitive rats. Hypertension. 2005; 45: 643-648.
- Michaelis UR, Fisslthaler B, Barbosa-Sicard E, Falck JR, Fleming I, Busse R. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J Cell Sci. 2005; 118: 5489-5498.
- Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). The FASEB Journal. 2003; 17: 770-772.
- Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007; 49: 590-596.
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999; 285: 1276-1279.
- Zhao G, Wang J, Xu X, Jing Y, Tu L, Li X, et al. Epoxyeicosatrienoic acids protect rat hearts against tumor necrosis factor-α-induced injury. J Lipid Res. 2012; 53: 456-466.
- Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 2007; 82: 50-59.
- Zhao G, Tu L, Li X, Yang S, Chen C, Xu X, et al. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum Gene Ther. 2012; 23: 688-699.
- Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, et al. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010; 24: 3770-3781.
- Yang S, Lin L, Chen JX, Lee CR, Seubert JM, Wang Y, et al. Cytochrome P-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-alpha via MAPK and PI3K/Akt signaling pathways. Am J Physiol Heart Circ Physiol. 2007; 293: 142-151.
- Liu Y, Webb HK, Fukushima H, Micheli J, Markova S, Olson JL, et al. Attenuation of Cisplatin-Induced Renal Injury by Inhibition of Soluble Epoxide Hydrolase Involves Nuclear Factor ? B Signaling. Journal of Pharmacology and Experimental Therapeutics. 2012; 341: 725-734.
- Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J. 2013; 27: 2946-2956.
- Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, et al. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond). 2009; 116: 61-70.
- Puntarulo S, Cederbaum AI. Production of reactive oxygen species by microsomes enriched in specific human cytochrome P450 enzymes. Free Radic Biol Med. 1998; 24: 1324-1330.
- Fleming I, Michaelis UR, Bredenkötter D, Fisslthaler B, Dehghani F, Brandes RP, et al. Endothelium-Derived Hyperpolarizing Factor Synthase (Cytochrome P450 2C9) Is a Functionally Significant Source of Reactive Oxygen Species in Coronary Arteries. Circ Res. 2001; 88: 44-51.
- Nilakantan V, Maenpaa C, Jia G, Roman RJ, Park F. 20-HETE-mediated cytotoxicity and apoptosis in ischemic kidney epithelial cells. Am J Physiol Renal Physiol. 2008; 294: 562-570.
- Baliga R, Zhang Z, Shah SV. Role of cytochrome P-450 in hydrogen peroxide-induced cytotoxicity to LLC-PK1 cells. Kidney Int. 1996; 50: 1118-1124.
- Zeng Q, Han Y, Bao Y, Li W, Li X, Shen X, et al. 20-HETE increases NADPH oxidase-derived ROS production and stimulates the L-type Ca2+ channel via a PKC-dependent mechanism in cardiomyocytes. American Journal of Physiology-Heart and Circulatory Physiology. 2010; 299: 1109-1117.
- Chen W, Zheng G, Yang S, Ping W, Fu X, Zhang N, et al. CYP2J2 and EETs Protect against Oxidative Stress and Apoptosis in Vivo and in Vitro Following Lung Ischemia/Reperfusion. Cell Physiol Biochem. 2014; 33: 1663-1680.
- Liu L, Chen C, Gong W, Li Y, Edin ML, Zeldin DC, et al. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. Journal of Pharmacology and Experimental Therapeutics. 2011; 339: 451-463.
- Hoagland KM, Maier KG, Roman RJ. Contributions of 20-HETE to the antihypertensive effects of Tempol in Dahl salt-sensitive rats. Hypertension. 2003; 41: 697-702.
- Enriquez A, Leclercq I, Farrell GC, Robertson G. Altered expression of hepatic CYP2E1 and CYP4A in obese, diabetic ob/ob mice, and fa/fa Zucker rats. Biochem Biophys Res Commun. 1999; 255: 300-306.
- Dey A, Maric C, Kaesemeyer WH, Zaharis CZ, Stewart J, Pollock JS, et al. Rofecoxib decreases renal injury in obese Zucker rats. Clin Sci (Lond). 2004; 107: 561-570.
- McCarthy ET, Sharma R, Sharma M. Protective effect of 20-hydroxyeicosatetraenoic acid (20-HETE) on glomerular protein permeability barrier. Kidney Int. 2005; 67: 152-156.
- Zhu Y, Usui HK, Sharma K. Regulation of transforming growth factor beta in diabetic nephropathy: implications for treatment. Semin Nephrol. 2007; 27: 153-160.
- Liang CJ, Ives HE, Yang CM, Ma YH. 20-HETE inhibits the proliferation of vascular smooth muscle cells via transforming growth factor-beta. J Lipid Res. 2008; 49: 66-73.
- Jung O, Jansen F, Mieth A, Barbosa-Sicard E, Pliquett RU, Babelova A, et al. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. Plos One. 2010; 5: 11979.
- Williams JM, Sharma M, Anjaiahh S, Falck JR, Roman RJ. Role of endogenous CYP450 metabolites of arachidonic acid in maintaining the glomerular protein permeability barrier. Am J Physiol Renal Physiol. 2007; 293: 501-505.
- Luo P, Zhou Y, Chang HH, Zhang J, Seki T, Wang CY, et al. Glomerular 20-HETE, EETs, and TGF-beta1 in diabetic nephropathy. Am J Physiol Renal Physiol. 2009; 296: 556-563.
- Chen G, Wang P, Zhao G, Xu G, Gruzdev A, Zeldin DC, et al. Cytochrome P450 epoxygenase CYP2J2 attenuates nephropathy in streptozotocin-induced diabetic mice. Prostaglandins Other Lipid Mediat. 2011; 96: 63-71.
- Minuz P, Jiang H, Fava C, Turolo L, Tacconelli S, Ricci M, et al. Altered release of cytochrome p450 metabolites of arachidonic acid in renovascular disease. Hypertension. 2008; 51: 1379-1385.
- Davydov DR. Microsomal monooxygenase in apoptosis: another target for cytochrome c signaling? Trends Biochem Sci. 2001; 26: 155-160.