
Review Article
J Endocr Disord. 2014;1(2): 1009.
A 21-Year Old Woman with Pheochromocytoma Associated with Hypercalcemia: A Case Report
Evgeny Farber, Nakhoul Nakhoul, Gershovich Maria, Ben-Itzhaq Ofer, Klouger Yoram and Nakhoul Farid*
Nephrology & Hypertension Division, Bar Ilan University, Israel
*Corresponding author: Farid Nakhoul, Nephrology and Hypertension Division, Baruch padeh poriya Medical center, Faculty of Medicine in Galilee, Bar Ilan University, Israel
Received: Aug 13, 2014; Accepted: Aug 30, 2014; Published: Aug 30, 2014
Abstract
We experienced a case of Pheochromocytoma (PCC), which was found in a 21-year-old woman with hypercalcemia. The leading symptom was abdominal pain and dysmenorrheal. Large ovarian cyst was diagnosed by ultrasound. During induction anesthesia for ovarian cystectomy, the woman developed tachycardia and severe hypertension. Her familial histories were unremarkable. Laboratory examinations showed hypercalcemia (12.5 mg/dl) which was diagnosed 3 years before her last admission to our hospital. Computed tomography showed a heterogeneous mass measuring 12 cm in the left adrenal gland, which had abnormal uptake with 123-I Metaiodobenzylguanidine scintigraphy (MIBG). Serum/urine catecholamines were highly elevated, and serum PTH was mildly increased. The patient underwent open abdominal surgery with En bloc surgical resection of PCC with left adrenalectomy and ovarian cystectomy. The tumor was histological diagnosed as typical pheochromocytoma. Blood pressure was normal with serum calcium slightly decreased after surgery. She was free of disease postoperatively after 6 months of follow up.
Keywords: Pheochromocytoma; Hypercalcemia; Ovarian Cyst
Introduction
Pheochromocytoma and paraganglioma represent the catecholamine-producing tumor arising from the chromaffin cells of the sympathetic ganglia in the adrenal medulla and extra-adrenal tissue, respectively [1-3]. These infrequent tumors are classically known to present with episodic hypertension, tachycardia, headache, and diaphoresis. Pheochromocytoma is diagnosed by elevated catecholamine and metanephrines in the serum/urine [4-6], and imaging studies including Computed Tomography scanning (CT) and 123-I Metaiodobenzylguanidine (MIBG) scintigraphy [7-9]. Although the majority of pheochromocytomas are sporadic/nonfamilial, a growing population of them has recently been found with involvement in familial genetic syndromes, such as multiple endocrine neoplasia type 2, von Hippel–Lindau disease, neurofibromatosis type 1, and paraganglioma syndrome types 1, 3 and 4 [11,12]. Although pheochromocytoma in childhood is rare, it represents the most common pediatric endocrine tumor.
The only definitive treatment consists of surgical resection. During manipulation of the tumor, dangerous amounts resulting in life-threatening events, including hypertensive crises, cardiac arrhythmias, myocardial infarction or ischemia, pulmonary edema and multiorgan failure [13,14]. Furthermore, the rapid decrease of catecholamine after surgery may result in severe hypotension. To prevent these life-threatening events from happening, preoperative management has been recommended. One of these therapies is the use of alpha-adrenoceptor blockers, which can counter the adrenergic effects of catecholamines. In addition, alpha- blockade permits intravascular volume expansion. Nowadays, some centers use the non-selective alpha-blocker phenoxybenzamine, while others use the selective alpha-blocker doxazosin [14,15]. A beta-blocker is often added to phenoxybenzamine therapy in order to decrease chronotropic activity.
Herein, we describe a case of active pheochromocytoma, which was found in a 21-year-old woman with persistent humeral hypocalcaemia, without any evidence of primary hyperparathyroidism by the routine screening. After adrenalectomy blood pressure was normalized but with persistent hypocalcaemia and high plasma PTH. Her family history was negative for arterial hypertension and syndromes [16].
Case Report
A 21-year-old woman with an unremarkable medical history, a part from taking hormonal treatment because of dysmenorrheal six month before admission. She denied any palpitations, chest pain or panic attacks. Her family history was negative for arterial hypertension. On 2010 she was diagnosed as suffering from hypocalciuric hypocalcaemia, but she was asymptomatic and no further investigation or follow up was done. She was admitted to the gynecological ring because of dysmenorrheal and abdominal pain after 3 month of hormonal treatment. Elective laparoscopic ovarian cystectomy by general anesthesia was recommended.
At admission blood pressure was 120 mmHg systolic and 80 mmHg diastolic with normal sinus rhythm of 72 beats/minute. Blood test showed plasma Creatinine of 0.7 mg/dl and normal electrolytes. Plasma calcium was 12 mg/dl with PTH of86 pg/ml. Tran abdominal sonography showed large ovarian cyst of 8cm.Chest X-Ray and normal electrocardiogram without left ventricular hypertrophy. Blood and urine tests are summarized in Table 1A &B. Serum level of 1, 25-dihydroxyvitamin D3 was normal.
![]()
Test
Value
Normal
Urinary creatinine
800 ug/gr /day
900-1200 mg/day
Metanephrines/creatinine
86 ug/gr/day
0–500� ug/gr/day
Normetanephrines/creatinine
9600 ug/gr/day
0-630 ug/gr/day
Epinephrine/creatinine
5.3 ug/gr/day
10-150 ug/gr/day
Norepinephrine/creatinine
850 ug/gr/day
24-92 ug/gr/day
Free Catecholamines Urine
855 ug/gr/day
42-250� ug/gr/day
Table 1: 24-hour urine values.
![]()
Test
Value
Normal
Glucose
98 mg/dl
70-110 mg/dl
BUN
8 mg/dl
7-17 mg.dl
Creatinine
0/5 mg/dl
0.6-1 mg/dl
Albumin
4.2 gr%
3-5� gr/dl
Potassium
4 meq/l
3.6-5 meq/l
Hb
13 gr
13-16 gr
Phosphorus
3.6 mg/dl
2.5-4.5 mg/dl
Calcium
11.6 mg/dl
8.5-10.5 mg/dl
Parathyroid hormone
86 pg/ml
12-65
TSH
1.95 miU/l
0.4-4/miU/l
Table 2: Blood test values.
A Computed Tomography (CT) scan of the abdomen and pelvis demonstrated a large left retroperitoneal mass (Figure 1) measuring 15/10/110 cm. The mass was predominantly cystic with irregular wall thickness and central necrosis. The mass was adherent to the left kidney and displaced it. There was no evidence of distant.
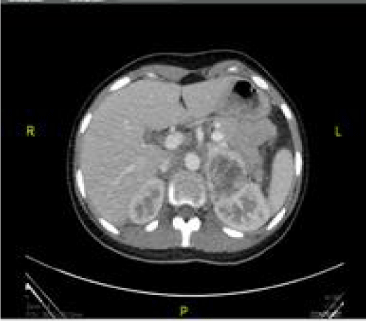
Figure 1: Abdominal CT scan showing a 15 X8 cms mass in left adrenal gland (Red Arrow) and displacing left kidney.
Metastases by CT scan. A 123-I Meta-Iodobenzylguanidine (MIBG) scan was arranged demonstrating avid uptake of radiotracer to the right retroperitoneal mass only (Figure 2). After a preoperative medical consult and adequate preoperative catecholamine blockade, the patient underwent left adrenal open adrenalectomies and ovarian cystectomy. The size of the adrenal mass consisting of pheochromocytoma was 10x15x15cm (Figure 3). Intraoperatively, there were no surgical complications; Adrenalectomy resulted in blood pressure normalization. She was discharged without any treatment. Histology confirmed a giant non-malignant pheochromocytoma PCC without capsular invasion or vascular invasion (Figure 4).
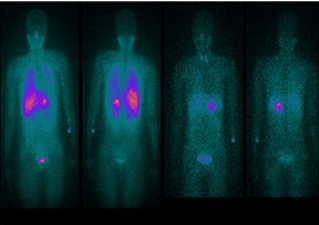
Figure 2: 123-1-MIBG scan of the patients: uptake in left adrenal pheochromocytoma.
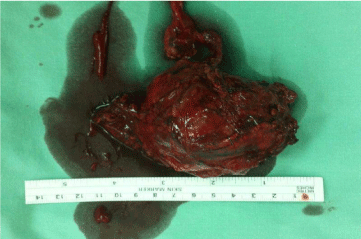
Figure 3: Macroscopic appearance of large circumscribed tan pheochromocytoma (By intraoperative open resection).
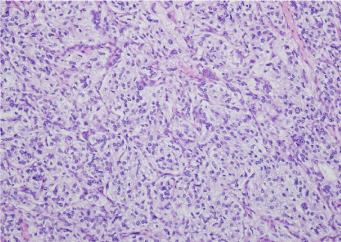
Figure 4A: Microscopic findings of the tumor with Hematoxylin–Eosin (HE) revealed a tumor proliferation of medium-size cells grouped into alveolar structures and solid nests; tumor cells presented round or oval nuclei.
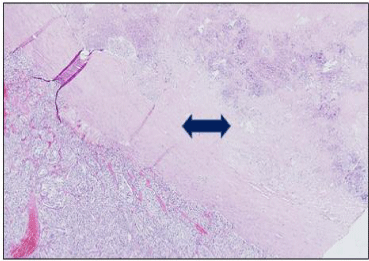
Figure 4B: Microscopic finding of the tumor showing central necrosis (blue arrow).
Because of persistent hypercalcemia and high Parathyroid Hormone (PTH) we performed a chest & neck CT scan with Sestamibi parathyroid scintigraphy without any evidence of parathyroid adenoma. Genetic consultation for RET testing is pending to consider multiple endocrine neoplasia type 2 (MEN type 2).
Discussion
PCC and Paraganglioma (PGL) are rare neuroendocrinetumors arising from chromaffin cells. PCCs are typically found in the adrenal medulla, accounting for 80%–85% of cases. Although Pheochromocytoma usually presents with classic symptoms, there is no single clinical sign or symptom specific for pheochromocytoma. Seventy-five percent of affected patients suffer from sudden and unexpected weekly attacks, while others just once every few months [1,2]. Hypertension is the most common sign, reported in 48% to 70% of cases, while dyspnea is only reported in 11% of cases [3]. PCC is diagnosed by measurements of catecholamine and metanephrines in the serum and in a 24-hour urine collection, with a 98% sensitivity and specificity [4-6]. A CT or magnetic resonance imaging of the abdomen is the imaging modalities of choice to localize a PCC, with a sensitivity of 90% to 100% and a specificity of 70% to 80%, respectively [6-8]. Hereditary PCC is of increasing interest because 10 genes are now known to play an important role in the pathogenesis of PCC. These genes include the RET7 (ret proto-oncogene), VHL (von Hippel- Lindau tumor suppressor, and NF1 (neurofibromin 1) tumor suppressor genes [11].
En bloc surgical resection is the standard treatment of PCC; it has 5-year survival rates of 95% for benign Pheochromocytoma. Essential intra-operative surgical steps include early isolation of the tumor’s venous drainage with minimal manipulation of the mass followed by complete resection of the tumor [12-14]. Preoperative management is essential to prevent hemodynamic instability and hypertensive crisis before or during surgery, which usually includes a-adrenergic blockade with ß-blockade, with sufficient hydration with intravenous fluids the night before surgery. Postoperatively, these patients must be monitored in the intensive care unit for at least 24 hours [15]. The size and location of the primary tumor were significant clinical risk factors for metastasis and decreased overall survival duration. These findings delineate the long term follow-up and treatment for these tumors. The follow-ups should include biochemical studies, such as plasma metanephrines, and radiographic studies, such as computed tomography or Magnetic Resonance Imaging (MRI) [16].
References
- Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Alì A, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000; 85: 637-644.
- Bravo EL. Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev. 1994; 15: 356-368..
- Mannelli M, Ianni L, Cilotti A, Conti A. Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol. 1999; 141: 619-624.
- Gerlo EA, Sevens C. Urinary and plasma catecholamines and urinary catecholamine metabolites in pheochromocytoma: diagnostic value in 19 cases. Clin Chem. 1994; 40: 250-256.
- Bravo EL, Tarazi RC, Gifford RW, Stewart BH. Circulating and urinary catecholamines in pheochromocytoma. Diagnostic and pathophysiologic implications. N Engl J Med. 1979; 301: 682-686.
- Raber W, Raffesberg W, Kmen E et al. Pheochromocytoma with normal urinary and plasma catecholamines but elevated plasma free metanephrines in a patient with adrenal incidentaloma. Endocrinologist. 2000; 10: 65-68.
- Bravo EL, Gifford RW. Current concepts. Pheochromocytoma: diagnosis, localization and management. N Engl J Med. 1984; 311: 1298-1303.
- Maurea S, Cuocolo A, Reynolds JC, Tumeh SS, Begley MG, Linehan WM, et al. Iodine-131-metaiodobenzylguanidine scintigraphy in preoperative and postoperative evaluation of paragangliomas: comparison with CT and MRI. J Nucl Med. 1993; 34: 173-179.
- Maurea S, Cuocolo A, Reynolds JC, Neumann RD, Salvatore M. Diagnostic imaging in patients with paragangliomas. Computed tomography, magnetic resonance and MIBG scintigraphy comparison. Q J Nucl Med. 1996; 40: 365-371.
- Eisenhofer G, Pacak K, Goldstein DS, Chen C, Shulkin B. 123I-MIBG scintigraphy of catecholamine systems: impediments to applications in clinical medicine. Eur J Nucl Med. 2000; 27: 611-612.
- Howe JR, Norton JA, Wells SA. Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: results of long-term follow-up. Surgery. 1993; 114: 1070-1077.
- Kimura S, Nishimura Y, Yamaguchi K, Nagasaki K, Shimada K, Uchida H. A case of pheochromocytoma producing parathyroid hormone-related protein and presenting with hypercalcemia. J Clin Endocrinol Metab. 1990; 70: 1559-1563.
- Walther MM, Keiser HR, Linehan WM. Pheochromocytoma: evaluation, diagnosis, and treatment. World J Urol. 1999; 17: 35-39.
- Shapiro B, Fig LM. Management of pheochromocytoma. Endocrinol Metab Clin North Am. 1989; 18: 443-481.
- Pullerits J, Ein S, Balfe JW. Anaesthesia for phaeochromocytoma. Can J Anaesth. 1988; 35: 526-534.
- Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T,et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011; 96: 717-725.