
Case Report
J Endocr Disord. 2014;1(2): 1010.
A Novel Mutation in the PYGM Gene Resulting in Mcardle Disease
Hui-Hui Wu1, Nai-Jia Liu1, Zi-Hui Tang3 and Jie Wen1,2*
1Department of Endocrinology and Metabolism, Fudan University, China
2Department of Endocrinology and Metabolism, Jing’an District Center Hospital of Shanghai, China
3Department of Endocrinology and Metabolism, Tongji University, Chi
*Corresponding author: Jie Wen, Departments of Endocrinology and Metabolism, Huashan Hospital of Fudan University, NO. 12 Wulumuqi Mid Road, Building 0#, Jing’an District, Shanghai, 200040
Received: July 11, 2014; Accepted: September 12, 2014; Published: September 15, 2014
Abstract
Mcardle disease, which is caused by an inherited deficit of myophosphorylase consequent to defects in the PYGM gene, is a pure autosomal recessive disorder. Official figures manifest that over-133 known mutations are associated with this disorder, only a minority of which have racial diversity. Here, we identified a novel mutation, 437T > C, in the case of a 70-year-old male patient with a lifelong history of fatigability, worsening on exertion. The presence of Mcardle disease was supported by a fresh muscle biopsy and confirmed by myophosphorylase activity assay. This possibly ethnicity-associated mutation may significantly facilitate the prediction and diagnosis of Mcardle disease in the region.
Keywords: Mcardle disease; Myophosphorylase deficiency; Mutation
Introduction
Mcardle disease, also known as type V glycogen storage disease, is the most common metabolic myopathy clinically characterized by exercise intolerance with premature fatigue, painful muscle cramps, and recurrent myoglobinuria triggered by static muscle contractions or dynamic exercise [1]. Symptoms typically appear in adolescence or early adulthood. Pathogenic mechanism of this autosomal recessive inherited condition is mutations in the gene (PYGM) encoding myophosphorylase C, the skeletal muscle isoform of glycogen phosphorylase, which catalyzes the phosphorolytic cleavage of glycogen to glucose-1-phosphate. Deficiency of myophosphorylase results in improper energy metabolize of individuals suffering from Mcardle disease, presented by glycogen deposition and energy deficit [2].
The human myophosphorylase gene (PYGM), located on chromosome 11q13 and made up of 20 exons, has been identified as the causative gene of Mcardle disease. Since mutations may involve almost every exon of the PYGM gene [3], the whole PYGM gene sequencing is generally required to identify pathogenic mutations. However, as most of the PYGM mutations are private, the possibility of finding new mutations has to be taken into account.
In the present study, we sequenced the whole PYGM exons to identify a novel mutation in a Chinese male patient, whose diagnosis was established on the basis of characteristic clinical history and confirmed by biochemical and genetic analysis.
Subject and Methods
Subject
The patient came to our observation was a 70-year-old man who complained of premature fatigue, exertional myalgia, and recurrent myoglobinuria triggered by strenuous activities. The symptoms affected his activity level on a daily basis since adolescence. He also experienced the “second wind” phenomenon, which is pathognomonic for Mcardle disease and occurs shortly after the initial myalgia/cramps [4].
Hematologic and laboratory examination
Examination of the routine hematologic parameters was performed, including the levels of lactate dehydrogenase and creatine phosphokinase, the evaluation of antinuclear antibody and immunologic factors (IgM and IgG), as well as the quantification of Ca2+, K+, Na+, creatinine and urea nitrogen which contain in serum.
A skeletal muscle biopsy with specific enzyme histo chemistry to demonstrate myophosphorylase deficiency is useful in establishing the diagnosis of this rare condition. The levels of lactic acid and pyruvic acid were also examined after a forearm ischemic exercise test and made a comparison with a normal control.
Molecular analysis
The DNA sample for genetic analysis was extracted from peripheral blood of the patient by standard method. Primers for the 20 exons of PYGM were designed by specific ABI Primer Designer (V3.0) on a basis of the DNA sequence from the gene bank (NCBI Reference Sequence: NG 013018.1). Each of the whole 20 exons of PYGM gene was amplified by polymerase chain reaction (PCR) from genome DNA. The PCR condition was 35 cycles of denaturation at 95OC for 30 seconds, annealing at 55OC for 30 seconds, and extension at 72OC for 50 seconds.
The reaction products were sequenced by an Applied Bio system 3730 automated DNA sequence and analyzed by the technelysium Chromas software (V2.3), the results of which were further blasted with the reported variants of PYGM gene in normal populations using the Variant Reporter software (V1.1) to determine whether the mutation we found is pathogenic.
Results
Laboratory evaluation revealed significant elevation in levels of lactate dehydrogenase (400 U/L, upper limit of normal is 220U/L ) and creatine phosphokinase (34 U, upper limit of normal is 30U ), which is common in patients of Mcardle disease. The negative result of antinuclear antibody test, along with the normal levels of IgM and IgG could help eliminate the possibility of autoimmune disease and infection. The content of Ca2+, K+, Na+, creatinine and urea nitrogen in serum was also in normal range. As well as the characteristic clinical history, the positive forearm ischemic exercise test which presented with a lower rise of lactic acid and pyruvic acid in comparison with that of control also strengthened the suspicion of Mcardle disease. On this basis, the diagnosis was further supported by the fresh biopsy which manifested a deficiency of glycogen phosphorylase activity in deltoid muscle from quantitation by enzyme assay and an elevation of non-membrane bound glycogen with electron microscopy.
Molecular analysis is preferred for confirmation because it does not require an invasive procedure and has advantage in terms of genetic consults. In the present study, sequencing of the whole PYGM exons discovered a novel 437T > C mutation in exon 1 (shown in Figure 1). The corresponding oligonucleotide primers employed for sequencing analysis was Forward: 5’ -GTG GAG TGA GGG CTG TGG- 3’, Reverse: 5’- CTG CAA TGG GAG GGT CTTG -3’.
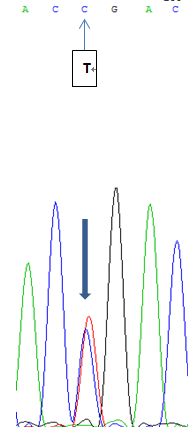
Figure 1: Overall#
Discussion
This is the first report of biochemically and genetically confirmed Mcardle disease in China, in spite of the cases diagnosed simply by clinical features and muscle biopsy findings in the region. According to previous reports, the onset of Mcardle disease varies from early/late childhood to adulthood and rare cases of late-onset forms (> 60 years) had been reported. In our study, the patient presented with signs of exercise intolerance such as myalgia and recurrent myoglobinuria triggered by exercise since adolescence, as a consequence of deficient myophosphorylase activity and block in glycogen breakdown. Performing an ischemic lactate test in someone with Mcardle disease poses some risks (contracture, rhabdomyolysis and compartment syndrome) and is thus not recommended. However, the patient was consent to perform a forearm ischemic exercise test, the result of which was a lower elevation of lactic acid and pyruvic acid levels than that of control, prompting a partial deficient of myophosphorylase. The sub sarcolemmal accumulation of glycogen could be appreciated on electron microscopy and the pathognomonic “second wind” phenomenon was also observed in the patient, energy requirement of muscle during which period may be supplied by glucose derived from hepatic glycogenolysis or by fatty acid oxidation (FAO).
Nearly all mutations in PYGM reduce or abolish the myophosphorylase activity and thus cause a partial or complete block of muscle glycogen breakdown. Therefore, individuals suffering from Mcardle disease can’t properly metabolize energy and exhibit exercise intolerance. Regardless of the growing allelic heterogeneity of the PYGM gene, most reported mutations, R50X and W798R, are specific ethnic mutations mainly identified in the patients from Western Europe, the United States and Japan [5-8], most of which are missense or nonsense mutations resulting in amino acid modification [9]. However, no mutation has been found in the promoter region of PYGM. At present, we first sequenced the whole 20 exons of PYGM gene and identified a novel mutation in a Chinese patient, 437T > C, which is located in the non-coding region of exon 1. The role of this PYGM polymorphism played in modulating phenotype remains a mystery. We speculate that it may be a promoter binding site which could induce an alteration in mRNA transcription or splicing, thus resulting to be pathogenic.
In spite of the enormous diversity among causative PYGM gene mutations, no genotype correlated with a distinct phenotype has been discovered [10]. Meanwhile, factors other than the single variants in the PYGM gene might influence the clinical presentation, for the different clinical manifestations presented by individuals with the same mutation [11]. Up to date, there is no definitive cure or therapeutic modalities for Mcardle’s disease. Simple measures of lifestyle modification and dietary changes benefit the patients [12].
Acknowledgement
The study was supported by the grant from National Natural Science foundation of China (81270903).
References
- Mcardle B. Myopathy due to a defect in muscle glycogen breakdown. Clin Sci. 1951; 10: 13-35.
- Haller RG, Vissing J. Spontaneous "second wind" and glucose-induced second "second wind" in McArdle disease: oxidative mechanisms. Arch Neurol. 2002; 59: 1395-1402.
- Andreu AL, Nogales-Gadea G, Cassandrini D, Arenas J, Bruno C. Mcardle disease: molecular genetic update. Acta Myol. 2007; 26: 53-57.
- Lucia A, Nogales-Gadea G, Pérez M, Martín MA, Andreu AL, Arenas J. Mcardle disease: what do neurologists need to know? Nat Clin Pract Neurol. 2008; 4: 568-577.
- Bruno C, Cassandrini D, Martinuzzi A, Toscano A, Moggio M, Morandi L, et al. Mcardle disease: the mutation spectrum of PYGM in a large Italian cohort. Hum Mutat. 2006; 27: 718.
- Aquaron R, Bergé-Lefranc JL, Pellissier JF, Montfort MF, Mayan M, Figarella-Branger D, et al, Molecular characterization of myophosphorylase deficiency (Mcardle disease) in 34 patients from Southern France: identification of 10 new mutations. Absence of genotype-phenotype correlation. Neuromuscul Disord. 2007; 17: 235-241.
- Vieitez I, Teijeira S, Fernandez JM, San Millan B, Miranda S, Ortolano S, et al. Molecular and clinical study of Mcardle's disease in a cohort of 123 European patients. Identification of 20 novel mutations. Neuromuscul Disord. 2011; 21: 817-823.
- Sugie H, Sugie Y, Ito M, Fukuda T, Nonaka I, Igarashi Y. Genetic analysis of Japanese patients with myophosphorylase deficiency (McArdle's disease): single-codon deletion in exon 17 is the predominant mutation. Clin Chim Acta. 1995; 236: 81-86.
- Deschauer M, Morgenroth A, Joshi PR, Gläser D, Chinnery PF, Aasly J, et al. Analysis of spectrum and frequencies of mutations in McArdle disease. Identification of 13 novel mutations. J Neurol. 2007; 254: 797-802.
- Bruno C, Cassandrini D, Martinuzzi A, Toscano A, Moggio M, Morandi L, et al. McArdle disease: the mutation spectrum of PYGM in a large Italian cohort. Hum Mutat. 2006; 27: 718.
- Dimaur S, Andreu AL, Bruno C, Hadjigeorgiou GM. Myophosphorylase deficiency (glycogenosis type V; McArdle disease). Curr Mol Med. 2002; 2: 189-196.
- Maté-Muñoz JL, Moran M, Pérez M, Chamorro-Viña C, Gómez-Gallego F, Santiago C, et al. Favorable responses to acute and chronic exercise in McArdle patients. Clin J Sport Med. 2007; 17: 297-303.