
Review Article
J Endocr Disord. 2016; 3(1): 1020.
The Role of Nervous System Glycogen during Hypoglycemia
Rockliffe AM¹, Ebling FJP and Brown AM1,2*
¹School of Life Sciences, University of Nottingham, UK
²Department of Neurology, University of Washington, USA
*Corresponding author: Brown AM, School of Life Sciences, University of Nottingham, Queens Medical Centre, Nottingham, NG7 2UH, UK
Received: December 18, 2015; Accepted: April 18, 2016; Published: April 21, 2016
Abstract
The mammalian brain contains glycogen but in concentrations much lower than in the liver and skeletal muscle, thus a role as a functional energy reserve has been dismissed. Glycogen in the central nervous system is located in astrocytes in the adult, and in the peripheral nervous system is expressed in myelinating Schwann cells. During periods of experimental aglycemia in rodent optic nerve, a model of central white matter, the stimulus evoked Compound Action Potential (CAP) is sustained for up to 30 minutes, and thereafter rapidly falls to zero. Optic nerve glycogen decreases during aglycemia and is exhausted after about 30 minutes. This temporal correlation between glycogen content and maintenance of the CAP suggests that in the face of aglycemia, glycogen supports conduction, but once the limited glycogen stores are exhausted the CAP fails. The glycogen is metabolised to lactate, which is shuttled from the astrocyte to the axon to serve as a transportable energy substrate. In the peripheral nervous system a similar scheme occurs in which Schwann cell glycogen supports conduction of large myelinated A fibres during aglycemia via. transfer of the glycogen-derived conduit lactate. The smaller unmyelinated C fibres do not benefit from the presence of glycogen. However inhibiting glycogen metabolism with DAB during aglycemia abolishes any benefit the A fibres derive from glycogen, and their latency to failure resembles that of the C fibres.
Keywords: Glycogen; Astrocyte; Hypoglycemia; Schwann cell; Isofagomine; DAB; D-lactate
Introduction
The human brain has an absolute reliance on blood borne glucose in order to function, although the actual energy substrate that the individual brain cells use may not be glucose, but a glucosederived substrate such as lactate. Endocrine functions are well developed to maintain systemic blood glucose levels such that the blood delivers glucose to the brain well in excess of demand. However certain pathological conditions such as insulinomas, and the iatrogenic consequences of mismatch between insulin delivery and prevailing systemic blood glucose levels in patients suffering from type 1 diabetes, can result in insufficient glucose delivery to the brain to support normal function. Under such conditions the brain suffers pathological consequences, which can range in severity from autonomic warning signals, such as trembling and hunger pangs for limited periods of hypoglycemia, but ultimately to death for extended periods of hypoglycemia. There are currently no clinically relevant neuroprotective therapies available to preserve brain tissue in the event of hypoglycemia.
Iatrogenic hypoglycemia
The normal blood glucose concentration varies between 4 and 7.2 mmol l-1 with complex endocrine functions responsible for maintaining this narrow normoglycemic range [1]. This regulation requires the actions of the complementary hormones insulin and glucagon, both released from the pancreas [1]. In times of plenty when glucose is abundant, insulin is released from the pancreatic beta cells and acts upon surface bound glucose transporters (Glut 4) to facilitate the transmembrane movement of glucose into cells [2], with the glucose incorporated into the glycogen macromolecule [2]. Glucagon acts in response to falling glucose levels and liberates glucose from glycogen storage in order to elevate blood glucose levels [3]. This intricate balance between glucose levels and hormone release is disrupted in type 1 diabetes, where autoimmune destruction of insulin secreting cells renders the sufferer unable to regulate glucose levels resulting in uncontrolled hyperglycemia [1]. Prior to the advent of insulin therapy in 1922 [4] a diagnosis of type 1 diabetes was an almost certain death sentence, but the clinical intervention of applying exogenous insulin is a very effective therapy, which if rigidly adhered to, allows patients to lead a relatively normal life [1]. However this therapy has one major drawback, namely the systemic hypoglycemia that starves the brain of glucose when insulin administration is mismatched to prevailing glucose levels [5]. The fear of such hypoglycemic episodes is the primary reasons sufferers of type 1 diabetes do not adhere strictly to such therapies, and is indirectly the cause of numerous pathologies, such a retinopathy, neuropathy and nephropathy, that result from persistent hyperglycemia [6].
Hypoglycemia symptoms
The effects of hypoglycemia can be broadly divided into two, autonomic symptoms and neuroglycopenic symptoms [1]. In the event of a hypoglycemic episode, as the blood glucose falls to levels below 4 mM, there occurs a reaction mediated by the autonomic nervous system whose symptoms include the following: sweating, trembling, difficulty concentrating, tenseness and light headedness, and dizziness [1]. Such symptoms are a warning of an impending hypoglycemic episode, which can be avoided if the patient rapidly ingests concentrated glucose in the form of a gel or high-energy drink. If no such interventions are made hypoglycemia can progress to evoke neuroglycopenic symptoms, which include confusion, drowsiness, unpredictable behaviour, speech difficulty and the loss of co-ordination [1]. Such symptoms may render the patient incapable of the reasoning required to take counter measures and as such are potentially life threatening. The ultimate pathology associated with hypoglycemia is neuronal death, which can occur relatively rapidly and encompass broad regions of the brain [7-9]. The autonomic warning symptoms at the onset of an impending hypoglycemic episode are complicated by the phenomenon of hypoglycemia unawareness. This is a condition in which the warning signs are missed by the patient for reasons as yet unknown. It is proposed that successive hypoglycemic episodes cause a pathological change to the autonomic warning signs, such that the threshold for the onset of these symptoms occurs at glucose concentrations lower than those that trigger the neuroglycopenic symptoms [6]. The pathology associated with hypoglycemia in the brain has been documented and consist of neuron death, with the intriguing aspect that some brain regions are more sensitive to hypoglycemic damage than others. However care must be taken to isolate the effects of hypoglycemia from those long-term effects of type 1 diabetes [8].
Glycogen in the central nervous system
The dogmatic view of brain energy metabolism is that there are no significant energy reserves within the brain [2], in the manner of glycogen in the liver or skeletal muscle, rendering the brain exquisitely sensitive to shortfalls in glucose delivery, and this view is supported by the following compelling evidence. Occlusion of the carotid artery renders human unconscious within 6 to 8 seconds [10], neuroglycopenic symptoms are temporally correlated with hypoglycemia [11], and no significant energy reserves have been located within the brain [2]. However, the brain must be assessed for the unique organ that it is, and assessment of its energy requirements accordingly, rather than comparison with organs such as the liver and skeletal muscle.
The most likely source of an energy supply in the brain, given its absolute reliance on glucose, is glycogen. Glycogen was first identified in the brain by biochemical assay [12], and later by electron microscopy, but interest in it as a functional entity was limited due to the very low concentration in which it occurs relative to the liver and skeletal muscle. The paucity of brain glycogen suggests that it does not play the role of an energy reserve in the manner of liver glycogen. To understand the role of brain glycogen we must carefully analyse the preliminary studies that led to suggestions of it as having an important role in energy metabolism, and these experiments concerned functional aspects of in vitro sections of brain exposed to hypoglycemia.
Effect of systemic hypoglycemia on brain glycogen
The advent of insulin therapy also introduced the spectre of hypoglycemia as a therapeutic treatment to alleviate symptoms of schizophrenia [13]. The widespread uptake of this practice demanded a more detailed knowledge of the effects of systemic hypoglycemia on brain function, and such studies commenced in the 1940s and 1950s. In a study on dogs, hypoglycemia provoked a decrease in glycogen content in areas of the brain recognized for their high metabolic rate, with the revealing detail being the depletion of glycogen depended on the metabolic rate of the region, and not the initial concentration [14]. Comparable studies in the rabbit showed a significant fall in glycogen in the brain after a period of hypoglycemia [15]. Collectively these studies suggested for the first time a link between brain glycogen content and brain function. Such basic biochemical studies have been supplemented with NMR spectroscopic data showing that insulininduced hypoglycemia is rats depletes glycogen content [16], results recently confirmed in human subjects [17,18]. Although a clear correlation between insulin induced hypoglycemia and brain glycogen has been established, the finer details of the brain glycogen metabolism required more invasive techniques, and these subsequent experiments have tended to be carried out in vitro. An initial study, although not intending to study any direct role of glycogen, highlighted the dependent nature between neuron survival and glycogen. Hypothalamic neurons that were co-cultured with astrocytes survived for longer periods than neurons cultured in isolation, linking the fate of neurons to the presence of astrocytes [19]. However it was not merely the presence of the astrocytes that sustained the neurons, as a subsequent study of cultured cortical cells demonstrated. Cortical neurons co-cultured with astrocytes in conditions designed to deplete glycogen showed decreased survival compared to neurons co-cultured with astrocytes with plentiful glycogen [20,21]. Thus the presence of glycogen was the key factor that promoted neuron survival. It is one of the key features of glycogen that it is located almost exclusively in astrocytes in the adult mammalian brain [22]. Such a cellular location dictates certain fundamental aspects of the role of glycogen, the most important of which is that there must be transport of energy substrate from the astrocyte to the neural (axon / neuron) element in order for the neuron to benefit from the presence of glycogen.
Glycogen supports neural function
The dominant experimental manoeuvre to study the role of glycogen during hypoglycaemia is to completely remove glucose from the tissue, i.e. expose the tissue to aglycemic conditions. Although such conditions are extreme and never occur even in the most extreme case of iatrogenic hypoglycemia in humans, it is a very useful protocol as it removes confusion as to whether glucose and / or glycogen is supporting function. In hippocampal slices of rat in which the stimulus evoked Excitatory Post Synaptic Potentials (EPSPs) from the CA1 region were recorded, EPSP slope was maintained in aCSF containing 10 mM glucose, but substitution with aCSF devoid of glucose led to delayed attenuation of the EPSP slope [23]. This delay could be decreased by pre-exposure of the hippocampal slice to conditions that would deplete the tissue of glycogen, prior to introduction of aglycemia [24]. In the rodent optic nerve model, a popular model of central white matter [25], the stimulus evoked Compound Action Potential (CAP) is a useful model of axon conduction, as it allows post insult area of the CAP to be compared to baseline to estimate the degree of injury incurred by the tissue [26]. Exposure of the Rat Optic Nerve (RON) to anoxic conditions resulted in a rapid fall of the CAP area to zero in less than 5 minutes [27]. However exposure of the RON to aglycemia led to a delayed failure of the CAP, up to 30 minutes, after introduction of aglycemia [28]. Such an extended latency to failure suggested that there was an endogenous energy reserve that could sustain function in the absence of glucose, but that the energy reserve was limited and was exhausted within 30 minutes, after which function could not be supported. This energy reserve is glycogen. From these preliminary findings the role of glycogen in the optic nerve was examined over the next decade, and its role under hypoglycemic and normal conditions was unravelled.
Based on the initial findings that in the absence of exogenously applied glucose the CAP was sustained for 30 minutes [28] the following hypothesis were developed, which could be tested experimentally.
- The glycogen content correlated with the presence of the CAP, with the disappearance of the CAP correlating with the exhaustion of glycogen.
- Down regulating the glycogen content prior to an aglycemic period would attenuate the latency to CAP failure.
- Up regulating the glycogen content prior to an aglycemic period would augment the latency to CAP failure.
These hypotheses were tested as follows in the RON. As previously described withdrawal of glucose from the optic nerve previously incubated in an aCSF containing 10 mM glucose causes the CAP to fail about 30 minutes after aglycemia introduction. A parallel set of experiments were carried out in which the RONs were exposed to aglycemia but harvested every 10 minutes, and the glycogen content assessed by biochemical assay. The basal level of glycogen was about 7 pmol μg protein-1, but this decreased over the period of aglycemia to the extent that glycogen content had fallen to 2 pmol μg protein-1 after 30 minutes of aglycemia, and from this point it fell no further. Some glucosyl molecules remain bound to the glycogenin skeleton, so although they can be measured biochemically they are non functional and thus 2 pmol μg protein-1 is equivalent to zero glycogen. The glycogen fell at a constant rate during aglycemia while the CAP was fully supported. However once the glycogen had reached its nadir, the CAP fell rapidly to zero [29]. Such a scenario suggests that during aglycemia glycogen metabolism is activated and the glycogen is broken down to support CAP function. However once all the glycogen has been exhausted there are no other energy reserves available to support the CAP and it fails rapidly.
Glycogen can be up or down regulated according to the glucose concentration the tissue is bathed in, thus incubating the nerve in high glucose (25 mM) results in elevated glycogen levels compared to control (10 mM) glucose. Glycogen can also be down regulated by incubating with 1 mM nor adrenaline, thus the glycogen content can be varied over a 3 fold range by incubating in 1 mM nor-adrenaline (lowest), 10 mM glucose (intermediate) or 25 mM glucose (highest). Incubating nerves in the above conditions prior to introduction of aglycemia led to differences in the latency to CAP failure, that are consistent with elevated glycogen content increasing the latency to CAP failure during subsequent aglycemia, and down regulating glycogen content leading to the opposite effect [29] (Figure 1).
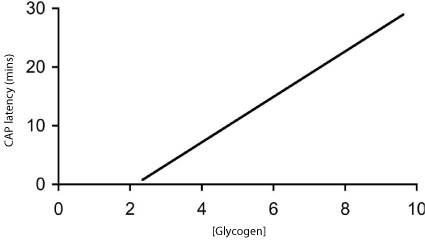
Figure 1: The relationship between glycogen content (pmol μg protein-1) at
the onset of glycemia and the latency to CAP failure in the MON.
Thus we have established a relationship between glycogen content and latency to CAP failure during subsequent aglycemia in the RON.
The implications of these results, when extrapolated to the human patient suffering from type 1 diabetes are profound. If a clinical intervention could be discovered that permitted up-regulation of brain glycogen levels, without any adverse side effects on either the brain, or on the general metabolic state of the individual, then such a manoeuvre could be used to stave off hypoglycemic episodes [30]. It is known that in a sufferer of type 1 diabetes one hypoglycemic episode tends to lead to another, so re-priming glycogen levels after one hypoglycemic episode may render the sufferer less susceptible to subsequent episodes.
Lactate as glycogen-derived conduit
Thus far we have discussed in general terms the correlation between the presence of glycogen and the RON conduction. However it is clear that since glycogen is a large macromolecule that can be visualised using electron microscopy, it is clearly unsuitable and unable to transfer between cells, thus some other glycogen-derived conduit must be available. The most likely candidate for this conduit is lactate. Under tissue culture conditions astrocytes release lactate in significant quantities [31]. Astrocytes do not express the enzyme glucose-6-phosphatase, thus they are unable to convert glucose-6- phosphate to glucose [32-34]. This means that once glucose enters astrocytes and is phosphorylated it is committed down a metabolic pathway that will not produce glucose. Thus astrocytes resemble skeletal muscle cells in this regard, whereas liver cells release glycogenderived substrate in the form of glucose into the systemic circulation. Recent recordings with lactate sensitive biosensors have convincingly shown sustained lactate release from both central [35] and peripheral tissue [36] in agreement with astrocytic release of lactate.
If lactate is the glycogen-derived conduit that is transported between astrocytes and axons the following hypotheses can be tested.
- Lactate, as sole exogenously applied energy substrate, should support the stimulus evoked CAP for extended periods of time.
- Interrupting the transfer of lactate from the astrocyte to the axon should accelerate latency of aglycemia induced CAP failure.
- Transporters selective for lactate (monocarboxylates transporters) should be present on both the astrocyte and axon cell membranes.
Since glucose has 6 carbon molecules and lactate has only 3, 20 mM lactate is the carbon equivalent of 10 mM glucose. Perfusing RONs with 20 mM lactate supported the CAP for periods in excess of 2 hours, confirming that lactate is transported across axon membranes by Monocarboxylates Transporters (MCTs) [29]. The compounds cinnemate and quercitin inhibit lactate transport thus these compounds were employed to dissect out astrocyte to axon metabolic signalling. Quercitin preferentially blocks extrusion of lactate from cells, whereas cinnemate blocks lactate uptake into cells. Quercitin had no effect when added in the presence of 10 mM glucose, which suggests that glucose is taken up directly into axons, rather than being shuttled via. the astrocyte and released as lactate. Similarly when added with 20 mM lactate as sole exogenously applied energy substrate quercitin had no effect on the CAP. However under aglycemic conditions quercitin accelerated the failure of the CAP indicating that preventing the releases of lactate from astrocytes accelerates CAP failure. Cinnemate, in the presence of 10 mM glucose, had no effect on the CAP, but in the presence of 20 mM lactate resulted in a partial failure of the CAP. Under aglycemic conditions cinnemate accelerated the CAP failure [22]. The combined data from the effects of quercitin and cinnemate strongly suggest that there occurs, during aglycemia, a uni-directional transfer of lactate from astrocytes to axons via. the extracellular space, a transfer that is mediated via. the actions of MCTs.
All subsequent studies reported employing the rodent optic nerve used the Mouse Optic Nerve (MON) as a model. The mouse is preferable over the rat for a variety of reasons that include: (1) decreased diffusional distance to the centre of the tissue, (2) compatibility with future transgenic and knock out models. The presence of MCTs in MON was investigated using antibodies directed against the MCTs in combination with cell specific markers. The MCT1 subtype of the monocarboxylates transporter family is expressed in tissue that releases lactate, whereas the MCT2 subtype is selectively expressed in tissue that takes up lactate. Staining for the MCT2 transporter was seen on neurofilament positive cells confirming an axonal expression of the MCT2, whereas the MCT1 transporter was seen on GFAP positive astrocytes, confirming the expression of a lactate-releasing transporter on astrocytes [37]. Thus, the cellular expression of these MCTs supports a uni-directional movement of lactate from astrocytes to axons.
Blockade of lactate transfer
In the MON, pharmacological block of the lactate uptake into axons was investigated using D-lactate, a metabolically inert but transportable monocarboxylate. D-lactate had no effect of the CAP in MONs perfused with 10 mM glucose, but accelerated CAP failure when applied at the onset of aglycemia. These results suggest that in MONs glucose is taken up directly into axons to act as energy substrate, since D-lactate had no effect on the glucose supported CAP. However the observation that D-lactate accelerated CAP failure during aglycemia support the uptake of glycogen-derived lactate into axons, to sustain CAP conduction in the absence of exogenously applied glucose. D-lactate applied in the presence of 2 mM glucose caused a failure of the CAP, whereas 2 mM glucose alone supported the CAP for periods of up to an hour [37]. Thus lactate appears to be transported to the axons in the presence of hypoglycemic concentrations of glucose to provide supplementary substrate. In addition, D-lactate attenuated CAP area during periods of 100 Hz stimulus in MONs supported by 10 mM glucose [37], again highlighting the transfer of glycogenderived lactate from astrocytes to axons to support the CAP when glucose delivery does not match the energy demands of the tissue (Figure 2).
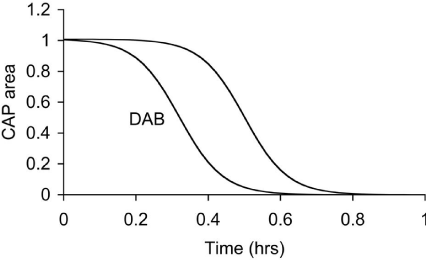
Figure 2: Latency to CAP failure in control MONs exposed to aglycemia, and
MONs exposed to aglycemia in the presence of DAB, an inhibitor of glycogen
phosphorylase.
Thus far the data support a scheme in which glycogen, located exclusively in astrocytes in the rodent optic nerve, is metabolized to lactate under conditions of aglycemia, hypoglycemia or increased tissue energy demand. The lactate is transported from the astrocyte to the axon via. MCTs, the lactate presumably being oxidatively metabolized by the axons. Additional evidence for such a scheme was sought using the compounds isofagomine and DAB that inhibit the breakdown of glycogen (glycogenolysis). Based on the data already presented some predictions can be made on the putative effects of these drugs.
- Isofagomine should accelerate CAP failure during a period of aglycemia.
- Isofagomine should attenuate the CAP during periods of high intensity stimulus.
The first prediction was tested under two conditions. Firstly, glucose was withdrawn from MONs previously bathed in 10 mM glucose. In MONs where isofagomine was applied during aglycemia the CAP latency to failure was decreased compared to control. In a second series of experiments MONs were pre-incubated in 30 mM glucose for 2 hours to up-regulate glycogen content prior to aglycemia. Addition of isofagomine resulted in accelerated failure of the CAP compared to MONs in which isofagomine was added. Isofagomine caused a decrease in the CAP area in MONs exposed to 100 Hz stimulus [38]. Thus, these data suggest there is mobilization of glycogen during periods of aglycemia, where glycogen-derived lactate is the sole energy substrate to support conduction, and during periods of increased metabolic demand where the normoglycemic concentrations of exogenously applied glucose are insufficient to fully support the CAP, and this glycogen-derived lactate provides supplemental substrate.
Peripheral nervous system glycogen
In addition to glycogen supporting axonal conduction in CNS tissue, glycogen also selectively supports axon conduction in the sciatic nerve, a peripheral nerve preparation, although there are significant differences when compared to the CNS. Whereas the central axons are myelinated by oligodendrocytes, in the peripheral nervous system axons are myelinated by Schwann cells [39]. The Schwann cells only ensheathe one axon, whereas in the CNS oligodendrocytes can myelinate up to 30 axons [39]. The peripheral nervous system contains no astrocytes, thus it is assumed that the functions carried out by astrocytes in the CNS are performed by the Schwann cells in the peripheral nervous system. As in the CNS, not all axons are myelinated, but such unmyelinated axons in the peripheral nervous system are enwrapped by Remak cells, which, although they do not myelinate the axons, wrap multiple axons within a bundle [40]. To date there has only been one study investigating the function of glycogen in peripheral nerves [36]. The enzymes that govern the formation and breakdown of glycogen, glycogen synthase and glycogen phosphorylase, respectively, are present in the sciatic nerve, although they were initially reported to be expressed exclusively in axons [41,42]. Such an axonal location for these enzymes contradicts the location of glycogen (and its associated enzymes) in the CNS where an exclusively astrocytic domain for glycogen exists. However it should be noted that glycogen is expressed in neurons in the pathological condition Lafora’s disease. The role of glycogen in sciatic nerves was studied using immuno histochemistry, electron microscopy, biochemical assay and electrophysiological recordings. Glycogen phosphorylase, the enzyme that breaks down glycogen, was localised in both Schwann cells and in axon cytoplasm. Electron microscopic studies were in agreement, showing that glycogen granules were located exclusively in the cytoplasm of the Schwann cell. No glycogen was located in either the axon cytoplasm of myelinated or unmyelinated axons, or in the cytoplasm of Remak cells. Such a glycogen location is intriguing as it suggests a similar role to that in the CNS, i.e. shuttling of glycogen-derived substrate between Schwann cells and axons. To test whether the glycogen had any functional role the stimulus evoked CAP was recorded. The CAP consists of twin peaks, an initial large A peak contributed to by large myelinated axons, and a slower smaller C peak, contributed to by small unmyelinated axons. On exposure to aglycemia i.e. withdrawal of glucose from the aCSF, the C peak began to fall after about 30 minutes and reached zero after about 3 hours, whereas the A peak was sustained for over 2 hours in the face of aglycemia and subsequently fell to zero after 5 hours. These extended periods relative to the central optic nerve suggest significant differences in glycogen content and / or metabolic rate (Figure 3). The glycogen content was found to be about 50% greater than the optic nerve content across a range of bathing glucose concentrations. Experiments where glycogen was measured every hour over the course of the aglycemic exposure showed that the glycogen fell from a baseline level of about 10 pmol μg protein-1 until it reached its nadir at about 2 hours, at which point the A peak fell towards zero. This temporal correlation between the failure of the A peak and glycogen exhaustion was investigated further. Sciatic nerves were pre-incubated in either 2, 10 or 30 mM glucose, which we have previously shown in optic nerve to alter glycogen content, high bathing glucose concentrations leading to higher glycogen content. Subsequent aglycemia led to a failure of the A peak that was dependent upon the glycogen content of the tissue. However such a relationship was not seen in the C fibres whose latency to failure was independent of glycogen content [36]. Thus it appears that A fibres are supported by glycogen during aglycemia, but unmyelinated C fibres are not. If this were the case then, based on optic nerve data, it is highly likely that glycogen is metabolised to lactate which is shuttled between Schwann cells and axons. In light of this lactate was added to the aCSF as the sole energy substrate and sustained the A peak for over 4 hours, but the C peak was only partially supported [36] (Figure 4). The presence of MCTs has recently been shown in large myelinated A fibres, but not in C fibres. Introducing the compound DAB, which is an inhibitor of glycogen phosphorylase, caused an attenuation of the latency to the failure of the A peak during aglycemia, such that its time course to failure was very similar to that of C fibres, from which we conclude that while glycogen sustains the A peak by providing glycogen-derived lactate during aglycemia, when the availability of glycogen is limited the A peak behaves in the manner similar to that of the C peak, which are not supported by glycogen [36].
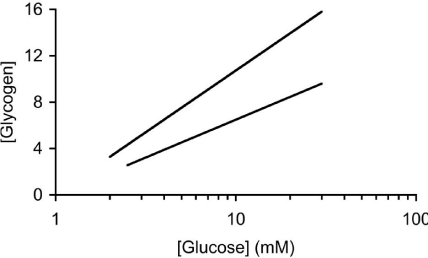
Figure 3: The sciatic nerve (steep line) contains more glycogen (pmol μg
protein-1) than the MON at equivalent [glucose].
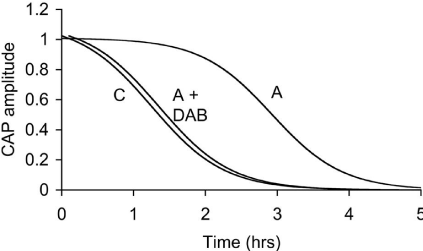
Figure 4: The latency to failure of the A peak of the sciatic nerve CAP is
significantly longer than that of the C peak. However incubating the sciatic
nerve in DAB, an inhibitor of glycogen phosphorylase, decreases the latency
to AS peak failure.
Hypothalamic glycogen
In addition to localization in astrocytes in the CNS, electron microscopy studies have revealed the presence of glycogen particles in hypothalamic tanycytes in rats and hamsters [43,44]. These observations are substantiated by in situ hybridization studies that have detected expression of key genes involved in glycogen metabolism in the ependymal cell layer containing tanycytes, for example glycogen phosphorylase [43]. Recent evidence suggests that tanycytes play a key role in nutrient sensing, glucose homeostasis and in adaptation to metabolic stress [45,46 for review], raising the question of whether glycogen of tanycyte origin plays a role in hypothalamic responses to hypoglycemia. A biological context in which this may be important is in adaption to winter torpor and hibernation, when the brain has to contend with relative hypoxia and hypoglycemia. Tanycytes display clear seasonal changes structure and function in seasonal mammals [47], and specifically glycogen phosphorylase mRNA abundance was observed to increase in tanycytes in hamsters exposed to winter photoperiodic conditions [43], mirroring previous observations of increased hypothalamic glycogen stores in hibernating hedgehogs in winter [48]. It is therefore tempting to hypothesize that tanycytes mobilize glycogen as a source of lactate to support neuronal function in hypothalamic neurons in periods of relative hypoglycemia during hibernation.
Conclusion
In both the peripheral and central nervous systems glycogen is present in glial cells, where it acts to supply glycogen-derived lactate to support axon conduction during periods of aglycemia. However, since glycogen reserves are limited the tissue benefits only for a short period of time. Although the introduction of aglycemia is a very useful experimental paradigm to study the functional role of glycogen in vitro, understanding the physiological role played by glycogen may be more rewarding from the point of view of normal function of the brain, since it will highlight metabolic signalling between glial cells and neural elements. How neurons signal to axons their energy requirements is a key topic that needs to be investigated, as does determining what is the contribution of glycogen under baseline and periods of increased metabolic activity.
Acknowledgments
FJPE’s contribution was supported by the Biotechnology and Biological Sciences Research Council (UK) project grants BB/ E020437/1 and BB/M001555/1.
References
- Frier BM, Fisher BM. Hypoglycaemia in Clinical Diabetes. New York: John Wiley and Sons, Ltd. 2007.
- Garrett RH, Grisham CM. Biochemistry. Fort Worth: Saunders College Publishing. 1999.
- Stryer L. Biochemistry. New York: W.H. Freeman Co. 1995.
- Tattersall RB. A force of magical activity: the introduction of insulin treatment in Britain 1922-1926. Diabet Med. 1995; 12: 739-755.
- Cryer PE. The pathophysiology of hypoglycaemia in diabetes. Diabetes Nutr Metab. 2002; 15: 330-333.
- Cryer PE. Hypoglycaemia: the limiting factor in the glycemic management of Type I and Type II diabetes. Diabetologia. 2002; 45: 937-948.
- Auer RN, Olsson Y, Siesjo BK. Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: a quantitative study. Diabetes. 1984; 33: 1090-1098.
- Auer RN. Progress review: hypoglycemic brain damage. Stroke. 1986; 17: 699-708.
- Auer RN. Hypoglycemic brain damage. Forensic Sci Int. 2004; 146: 105-110.
- Rossen R, Kabat H, Anderson JP. Acute arrest of cerebral circulation in man. Arch Neurol Psych. 1943; 50: 510-528.
- Amiel SA. Cognitive function testing in studies of acute hypoglycaemia: rights and wrongs? Diabetologia. 1998; 41: 713-719.
- Champe PC, Harvey RA. Biochemistry. Baltimore: Lippincott Williams & Wilkins. 2008.
- Rinkel M, Himwich HE. Insulin Treatment in Psychiatry. New York: Philosophical Library Inc. 1959.
- Chesler A, Himwich HE. Effect of insulin hypoglycaemia on glycogen content of parts of the central nervous system of the dog. Arch Neurol Psychiat. 1944; 52: 114-116.
- Goncharova ED. Effect of insulin and adrenalin on the polysaccharide metabolism in the brain. Reports of the Acad Sci, Ukranian USSR. 1957; 183-185.
- Choi IY, Gruetter R. Regulation of rat brain glycogen metabolism in vivo. J Neurochem. 2001; 78.
- Oz G, Henry PG, Seaquist ER, Gruetter R. Direct, noninvasive measurement of brain glycogen metabolism in humans. Neurochem Int. 2003; 43: 323-329.
- Oz G, Seaquist ER, Kumar A, Criego AB, Benedict LE, Rao JP, et al. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am J Physiol Endocrinol Metab. 2007; 292: 946-951.
- Whatley SA, Hall C, Lim L. Hypothalamic neurons in dissociated cell culture: the mechanism of increased survival times in the presence of non-neuronal cells. J Neurochem. 1981; 36: 2052-2056.
- Swanson RA. Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can J Physiol Pharmacol. 1992; 70: 138-144.
- Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993; 13: 162-169.
- Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. I Neurons and glia J Elec Micro Tech. 1986; 3: 413-437.
- Izumi Y, Benz AM, Katsuki H, Zorumski CF. Endogenous monocarboxylates sustain hippocampal synaptic function and morphological integrity during energy deprivation. J Neurosci. 1997; 17: 9448-9457.
- Cater HL, Benham CD, Sundstrom LE. Neuroprotective role of monocarboxylate transport during glucose deprivation in slice cultures of rat hippocampus. J Physiol. 2001; 531: 459-466.
- Ransom BR, Waxman SG, Fern R. Pathophysiology of white matter anoxic injury. In Cerebrovascular Disease. Lippincott Raven. 1997. 309-318.
- Stys PK, Ransom BR, Waxman SG. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 1991; 546: 18-32.
- Stys PK, Waxman SG, Ransom BR. Na+-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol. 1991; 30: 375-380.
- Ransom BR, Fern R. Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia. 1997; 21: 134- 141.
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci. 2000; 20: 6804-6810.
- Swanson RA, Bergher JP, Suh SW, Anderson CM. Manipulation of brain glycogen levels in vivo: effects on neuron function and survival during severe hypoglycemia. J Neurochem. 2005; 94: W04-02.
- Dringen R, Peters H, Wiesinger H, Hamprecht B. Lactate transport in cultured glial cells. Dev Neurosci. 1995; 17: 63-69.
- Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 1993; 623: 208- 214.
- Magistretti PJ, Sorg O, Martin J-L. Regulation of glycogen metabolism in astrocytes: physiological, pharmacological, and pathological aspects. In Astrocytes: Pharmacology and Function. Academic Press, Inc. 1993. 243- 265.
- Gotoh J, Itoh Y, Kuang TY, Cook M, Law MJ, Sokoloff L. Negligible glucose- 6-phosphatase activity in cultured astroglia. J Neurochem. 2000; 74: 1400- 1408.
- Yang X, Hamner MA, Brown AM, Evans RD, Ye Z, Chen SD, et al. Novel hypoglycemic injury mechanism: N-Methyl-D-Aspartate receptor–mediated white matter damage. Ann Neurol. 2014; 75: 492-507.
- Brown AM, Evans RD, Black J, Ransom BR. Schwann cell glycogen selectively supports myelinated axon function. Ann Neurol. 2012; 72: 406- 418.
- Tekkok SB, Brown AM, Westenbroek R, Pellerin L, Ransom BR. Transfer of glycogen-derived lactate from astrocytes to axons via. specific monocarboxylate transporters support mouse optic nerve activity. J Neurosci Res. 2005; 81: 644-652.
- Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, et al. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. 2005; 79: 74-80.
- Verkhratsky A, Butt A. Glial Neurobiology. Wiley and Sons. 2007.
- Peters A, Palay SL. Fine structure of the Nervous System: The Neurons and Supporting Cells. Philadelphia: W.B. Saunders Company. 1976.
- Pfeiffer-Guglielmi B, Fleckenstein B, Jung G, Hamprecht B. Immunocytochemical localization of glycogen phosphorylase isozymes in rat nervous tissues by using isozyme-specific antibodies. J Neurochem. 2003; 85: 73-81.
- Pfeiffer-Guglielmi B, Francke M, Reichenbach A, Hamprecht B. Glycogen phosphorylase isozymes and energy metabolism in the rat peripheral nervous system--an immunocytochemical study. Brain Res. 2007; 1136: 20-27.
- Nilaweera KN, Herwig A, Bolborea M, Campbell G, Mayer CD, Morgan PJ, et al. Photoperiodic regulation of glycogen metabolism, glycolysis, and glutamine synthesis in tanycytes of the Siberian hamster suggests novel roles of tanycytes in hypothalamic function. Glia. 2011; 59: 1695-1705.
- Lima SS, Lima Dos Santos MC, Sinder MP, Moura AS, Barradas PC, Tenorio F. Glycogen stores are impaired in hypothalamic nuclei of rats malnourished during early life. Nutritional Neuroscience. 2010; 13: 21-28.
- Bolborea M, Dale N. Hypothalamic tanycytes: potential roles in the control of feeding and energy balance. Trends in Neurosciences. 2013; 36: 91-100.
- Langlet F. Tanycytes: a gateway to the metabolic hypothalamus. Journal of Neuroendocrinology. 2014; 25: 753-760.
- Ebling FJ. Hypothalamic control of seasonal changes in food intake and body weight. Frontiers in Neuroendocrinology. 2015; 37: 97-107.
- Wittkowski W, Mueller K. Researches on the infundibulum of the hedgehog. Verhandlungen der Anatomischen Gesellschaft. 1976; 70: 49–54.