
Review Article
J Endocr Disord. 2020; 6(1): 1032.
Cellular Transporter Proteins in Cholesterol Metabolism
Sozmen EY1* and Sozmen B2
¹Department of Medical Biochemistry, Ege University, Turkey
²Department of Internal Medicine, Izmir University, Turkey
*Corresponding author: Eser Yildirim Sözmen, Ege University, Department of Medical Biochemistry, Turkey
Received: January 09, 2020; Accepted: February 19, 2020; Published: February 26, 2020
Abstract
Cholesterol from sources both endogenous and exogenous is delivered by plasma membrane receptors via endocytosis. While ApoB receptors and LDL receptors bind LDL cholesterol outside of the membrane, LDL receptor adaptor proteins control these processes. Lysosomal acid lipase enzyme hydrolyzes ester cholesterol into free cholesterol in lysosome. Lysosomal cholesterol is transported to the endoplasmic reticulum, the plasma membrane, mitochondria and/or peroxisomes through integral membrane proteins (NPC-1, NPC-2) Steroidogenic Acute Regulatory Protein Related Lipid Transfer Domain-3 (STARD3), Ras superfamily and OSBP Related Proteins, (ORPs). Free cholesterol is exported by ABCA1 and ABCG1 receptors. In this mini review, all these proteins affecting cellular transport and metabolism of cholesterol will be discussed and the diseases related to deficiencies and/or defects of these proteins will be summarized. De novo synthesis and control of cholesterol metabolism are out of focus of this review.
Keywords: Cholesterol; NPC1; NPC2
Abbreviations
ABCA1: Adenosine Triphosphate-Binding Cassette Transporter; A1; ACAT: Acyl-CoA Cholesteryl Acyl Transferase; ARH: Autosomal Recessive Hypercholesterolaemia; CESD: Cholesteryl Ester Storage Disease; EGF: Epidermal Growth Factor; ER: Endoplasmic Reticulum; ESCRT: Endosomal Sorting Complex Required For Transport; HDL: High Density Lipoprotein; HMGCR3: Hydroxy 3 Methylglutaryl Coa Reductase; LAL: Lysosomal Acid Lipase Enzyme; LAMP1: Lysosomal- Associated Membrane Protein 1; LDL: Low Density Lipoprotein; LDL: RAP1LDL Receptor Adaptor Protein1; LDL: LDL Receptor Family; LE: Late Endosome; LRP1: Alpha2 Macroglobulin Receptor or CD91; LY: Lysosome; MLN64: Endosomal Metastatic Lymph Node Protein 64; NPC1: Niemann Pick Type 1 Protein; NPC2: Niemann Pick Type C2 Protein; ORDOSBP Related Domain; ORPs OSBP: Related Proteins; ORPs OSBP: Related Proteins; OSBP: Oxysterol Binding Protein; PCSK9 Proprotein Convertase Subtilisin /Kexin Type 9; STARD3NLSTARD3 N Terminal like; STARD3 Steroidogenic Acute; Regulatory Protein Related Lipid Transfer Domain 3; START: Steroidogenic Acute Regulatory Protein; VAPVAMP Associated Protein; VLDL: Very Low Density Lipoprotein; VSMC: Vascular Smooth Muscle Cell.
Introduction
Intracellular Cholesterol transport is accomplished by two main roats: vesicular and non-vesicular. Vesicular transport requires an intact cytoskeleton where cholesterol moves along cytoskeletal proteins via energy supplied by dephosphorylation of ATP. Non vesicular transport requires carrier proteins which are the subject of this review.
Cellular cholesterol is transported by various membrane proteins and free transport proteins inside cell. After endocytosis of lipoproteins, cholesteryl esters are hydrolyzed by acid lipases in lysosomes. Free cholesterol might be transported to endoplasmic reticulum, plasma membrane, mitochondria and/or peroxisome by transport proteins [1]. This process and the transport proteins which have a role in this pathway will be discussed in following order.
A. Cellular uptake of Cholesterol via endocytosis of low density lipoproteins
B. Metabolism of ester cholesterol in Lysosome; Lysosomal Acid Lipase
C. Intracellular Trafficking of Cholesterol
D. Cellular Removal of Cholesterol
A. Cellular uptake of cholesterol via endocytosis of low density lipoproteins
Cellular uptake of cholesterol via endocytosis of low density lipoproteins is achieved by collaboration of various proteins including ApoB receptor, LDL Receptor family (LDLR), LDL Receptor Adaptor Protein-1 (LDL RAP-1), Proprotein Convertase Subtilisin /Kexin Type 9 (PCSK9), and Sortilin-1.
The members of LDLR family especially; LDLR, VLDLR, LRP5/6, LRP1, and LRP2 (megalin) participate in cholesterol homeostasis and lipid metabolism. Their structure is similar to common motifs, LDLR type A repeats, Epidermal Growth Factor (EGF)-like domain, transmembrane anchor, and cytoplasmic domain. LDLR recognizes and binds apoB-100 of LDL particles VLDL, IDL, High-Density Lipoprotein (HDL), and chylomicron remnant at neutral pH [2]. LDL particles play a vital role in cholesterol homeostasis inside of cell in terms of inhibiting the gene expression of 3-Hydroxy- 3-Methylglutaryl-Coa Reductase (HMGCR), stimulating Acyl- Coa Cholesteryl Acyl Transferase (ACAT) and suppressing LDLR synthesis to reduce LDL uptake [2].
After binding of one apoB-100 LDL particle with one LDL receptor monomer, this complex undergoes endocytosis. Apo-E containing lipoproteins are also recognized and bound by VLDLR, LDLR, LRP8, LRP1, LRP2, and LRP6. While VLDLR is expressed in adipose tissue, skeletal muscle, heart, endothelial cells of capillaries, and small arterioles, LDLR is expressed in hepatocytes and LRP8 (ApoER2) in brain, testis, and placenta [2].
![]()
GENE
DISEASE
FREQUENCY
METABOLIC DEFECT
LDLR
FH
1/400
Lack of function of LDL receptor Elevated LDL cholesterol
ApoB 100
FDB
1/800
LDLR:ApoB binding problem-Elevated LDL cholesterol
PCSK9 proprotein convertase subtilisin-like kexin type 9
FH3
1/2500
Increasing LDLR destroying Elevated LDL cholesterol
LDLRAP1 LDLR adaptor proteiARHn 1
1/5 000 000
Defect in uptake of LDLR into cell Elevated LDL cholesterol
NPC1,NPC2 Niemann Pick Type C protein
Niemann Pick Type C
1/100 000*
Defect in intracellular trafficking of cholesterol Elevated LDL cholesterol and ester cholesterol
LAMP2 Lysosome associated membrane protein
CESD WD
1/130 000 1/40 000**
Defect in hydrolysis of lipoprotein binding cholesterol ester and triglyceride Elevated levels of Ester cholesterol and triglyceride Type IIb dyslipidemia
ABCA1
Familial HDL deficiency Tangier disease
<1/ 1 000 000
Low levels of HDL cholesterol and apoprotein A-I. Cholesteryl esters accumulate in macrophage-rich tissues
ABCG5 and ABCG8 (encoding sterolin-1 and -2)
Sitosterolemia
Normal/mildly elevated levels of cholesterol.High levels of plant sterols
Table 1: The diseases related to lack/deficiency of proteins involved in cholesterol metabolism in cell. (https://www.orpha.net/consor/cgi-bin/Disease_Search. php?lng=EN ) *Alobaidy H, Schulz ML 2011. ARH: Autosomal Recessive Hypercholesterolaemia; CE: Cholesterol Esters; CESD: Cholesteryl Ester Storage Disease; FDB: Familial Defective Apolipoprotein B; FH: Familial Hypercholesterolaemia; FH3: Familial Hypercholesterolaemia 3; LDLR: LDL receptor; LDLRAP1, LDLR Adaptor Protein 1; LIPA: Lysosomal Acid Lipase A; PCSK9: Proprotein Convertase Subtilisin-Like Kexin Type 9; WD: Wolman Disease.
LRP1 (alpha2-macroglobulin receptor or CD91) is a receptor for more than 40 ligands including apoE-rich lipoprotein and is expressed in the liver and brain. LRP1 regulates the activities of proteases and protease inhibitor complexes and it has been shown that LRP1 has an essential role in protecting against atherosclerosis by reducing Vascular Smooth Muscle Cell (VSMC) proliferation and regulating levels of PDGF receptor in the vessel wall [2].
LRP2 (megalin) is a receptor for lipoprotein, steroid, and retinoid and is expressed in epithelial cell types. LRP2 has a role in endocytosis of HDL cholesterol [5] via binding with cubilin.
LRP6/LRP5 complex is co-receptor for Wnt/β-catenin signaling. LRP6 has a main role in LDL clearance by forming a complex with clathrin. It has been shown that a missense mutation in LRP6 has closely associated with early onset of cardiovascular disease and metabolic syndrome traits [2,3].
Sortilin-1 is a cell-surface receptor protein for LDL and it has been suggested that it mediated cellular uptake and degradation of LDL [4]. Sortilin-1 and PCSK9 co-localize in the Golgi and Sortilin-1 facilitates PCSK9 secretion from hepatocytes [4]. LDL RAP-1 (LDL Receptor Adaptor Protein-1) mediates internalization of the LDLR: LDL(ApoB): PCSK9 complex which is then delivered to the endosomes [5].
The mutations in the gene encoding LDLRAP1 (MIM #605747) result in Autosomal recessive hypercholesterolaemia (ARH) which is rare type of hypercholesterolemia (1/5 000 000 and it has mild clinical findings. The Mutations in the genes of LDLR (MIM #606945), apoB (MIM #107730) and PCSK9 (MIM #607786) result in Familial Hypercholesterolemia, Familial defective ApoB and Familial Hypercholesterolemia-3 respectively which are autosomal dominant and more common than ARH. (Prevalance is 1/500 of ADH and 1/5 000 000 of ARH). [5,6]. ARH prevalence: Familial hypercholesterolemia is classified into 5 groups regarding the mutations in LDLR proteins: 1) receptor synthesis-defect; 2) defect in transport alleles; targeting receptor to cell surface; 3) defect in binding ligands; 4) defect in internalization with clathrin coated pit; and 5) defect in recycling [2].
B. Metabolism of ester cholesterol in lysosome; lysosomal acid lipase
Lysosomal Acid Lipase Enzyme (LAL, EC 3.1.13) (lipase A, Acid Cholesteryl Ester Hydrolase, Acid Cholesterol Esterase, and Acid Cholesteryl Esterase) breaks down ester bonds in cholesteryl esters and triglycerides via hydrolysis and facilitates efflux of free fatty acid and free cholesterol from lysosome. LAL enzyme is coded by (LIPA) and the mutations in this gene resulting in LAL deficiency related diseases; infantile-onset, Wolman Disease (WD) (MIM 278000) and later-onset, Cholesteryl Ester Storage Disease (CESD). Cholesterol and tryglyceride accumulation in lysosomes of liver cells, adrenal glands, intestines and the macrophages/monocytes, lead to clinical manifestations such as atherosclerosis, stroke, fatty liver, etc. [7-9]. Wolman disease patients have no LAL activity and most of them die in their first year of life due to hepatic and adrenal failure [5]. Since CESD patients have residual LAL activity their clinical findings from medium hypercholesterolemia to hepatic failure regarding to enzyme activity [6,9]. More than 40 LIPA mutations which cause CESD and WD have been identified till now, [7]. Elevated cholesterol synthesis due to the lack of feed-back inhibition of 3- Hydroxy-3-Methylglutaryl Coenzyme A (HMG-CoA) reductase enzyme and the upregulation of apolipoprotein B (Apo B) synthesis and LDL-receptors on cell membranes lead to high levels of serum triglycerides, serum total and LDL cholesterol and down regulation of expression of the adenosine triphosphate-binding cassette transporter A1 (ABCA1) lead to reduced formation of mature HDL (a-HDL particles) and then low levels of serum HDL-cholesterol [7,9,10]. In normal cells, nuclear Liver X-receptor (LXR), is activated by increased cell cholesterol content and acts on the promoter of the ABCA1 gene. Since cholesterol accumulated in lysosomes in LAL deficient cells, intracellular free cholesterol levels decrease and ABCA1 mediated transfer of APO-A1 is diminished [9]. VLDLR up-regulates Lippoprotein Lipase (LPL) and increases uptake of Triglyceride-rich lipoproteins (chylomicron and VLDL) in endothelial cell as well as extrahepatic tissues such as heart and brain [2].
C. Intracellular trafficking of cholesterol
Free cholesterol in late endosome/lysosome is transferred to the other compartments of cell. The main proteins which are responsible for cholesterol efflux from lysosome are NPC1 and NPC2 proteins. NPC1 with 13 transmembrane domain is localized in lysosome membrane, NPC2 is 151-amino acid glycoprotein and soluble in lysosomal lumen. It has been suggested that NPC2 Protein binds free cholesterol in lysosomal lumen and transfers it to the NPC1 in membrane [11-13]. The luminal loops of NPC1 protein have distinct roles in cholesterol efflux; Loop1 is N Terminal Domain (NTD), can bind cholesterol, Loop2 binds NPC2 [12,13]. The Sterol Sensing Domain (SSD) between Loop 2 and Loop3 also can bind cholesterol [13]. The primary mutations of these proteins might lead to NPC disease which is characterized by accumulation of free cholesterol and other lipids in perinuclear lysosomes. More than 200 mutations have been determined in NPC1gene till now and approximately half of these mutations are localized to third loop of NPC1 protein [12-15]. Niemann-Pick disease type C (NP- C) which is a lysosomal storage disease with an incidence 1 in 120,000 live births, it’s caused by mutation in the genes NPC1 (95% of cases) or NPC2 (≈4% of cases). This autosomal recessive disease which is characterized by accumulation of cholesterol and glycosphingolipids in various tissues (brain, liver and spleen) due to impairment of cellular trafficking of LDL-derived cholesterol and other lipids. The clinical manifestations change regarding to the organs (brain, liver and spleen) in which accumulated lipids.
Many proteins having sterol-sensing domains on the cytosolic face of late endosomes have been identified till now. The main sterol carrier proteins in cytosol are Oxysterol Binding Protein (OSBP) and its relatives (OSBP related proteins, ORPs) [12,16]. ORPs, consisted of 12 members in human, have an OSBP-Related Domain (ORD) for binding of cholesterol, oxysterols and phosphatidylinositol 4-phosphate [17]. ORD with has capability of binding two different organelle membranes simultaneously and, plays a role in regulation of sterol transfer between the contact sites in terms of ER to LE/ LY or plasma membrane to ER [3,18-20]. Especially ORP5 forming a transient protein complex with NPC1 at the membrane contact sites, leads to transport of cholesterol from LE/LY directly to ER [12]. ORP5, is one of the Oxysterol Binding Protein-Related Proteins (ORP proteins), bounded to endoplasmic reticulum membrane and transfers sterols especially cholesterol between endoplasmic reticulum and late endosomes by interacting with NPC1 [21]. It has been shown that ORP5 and NPC1 worked together in transfer of cholesterol in late endosomal/lysosomal membranes of ORP5 knock down and also ORP5/NPC1 double knockdown cells [21]. ORP5 protein can also interact with Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) which regulates endosomal cholesterol transport via an independent mechanism from NPC2 and NPC1 [3]. Du X et al reported that Hrs played an essential role in the transport of LDL-C to the ER [3]. The role of Hrs on endosomal cholesterol trafficking might be explained in two ways; 1) Hrs mediates intraluminal vesicles for trafficking of cholesterol within endosomal lumen 2) Hrs facilitates the removal of cholesterol via cytoplasmic carriers (e.g.ORP5) [3].
The Endosomal Sorting Complex Required For Transport (ESCRT) is a hetero oligomeric complex protein, which consists of four protein complexes named as following 0-1-2 and 3. ESCRT Complex-0 is comprised from Hrs (Hepatocyte Growth Factor- Regulated Tyrosine Kinase Substrate) and STAM (Signal Transducing Adaptor Molecule), has a main role in transport of cholesterol from endosomes to the endoplasmic reticulum. It has been shown that the other complexes (1, 2, and 3) have no role in cholesterol trafficking in the cell [3].
Synthesized Cholesterol might be transferred from ER to plasma membrane through vesicular transport system mainly protein secretory pathway in golgi [22].
Significant amount of LDL-C is transferred via vesicular trafficking from NPC1-containing endosomal compartment to Trans Golgi Network [23]. The Ras-superfamily especially Rab8 and Rab9 proteins play main role in the vesicular trafficking. Rab8 and Rab9, small GTPase molecules, are released from membranes loaded with cholesterol. While Rab8 plays a role in transport of cholesterol from late endosomes to the cell surface via ABCA1, Rab9 plays a role in transport of cholesterol from late endosomes to the trans-Golgi network [23]. Both protein expression levels were found as increased in NPC1-deficient fibroblasts, so that the transport of cholesterol from LE/LY to plasma membrane is restored and cellular free cholesterol levels decreased [12,24]. The complex of RAB7, RAB7-Interacting Lysosomal Protein (RILP), and ORP1L regulates recruitment of the dynein motor and vacuole protein sorting (HOPS) complex to late endosomes. ORP1L has a role in transfer of cholesterol out of the LE, by inducing contact sites between LE and the ER, binding of the dynein motor and the HOPS complex to RAB7-RILP. Neuronal Ceroid Lipofuscinosis Protein (CLN3) associating with the RAB7- RILP complex also controls LE transport. Mutation in this protein causes classical juvenile onset Neuronal Ceroid Lipofuscinosis (NCL3, Batten (Spielmeyer-Vogt-Sjögren, CLN3), a fatal inherited neuro-degenerative lysosomal storage disorder.
Transport between lysosome and endoplasmic reticulum is achieved via proteins such as Steroidogenic Acute Regulatory Protein (START), Steroidogenic acute regulatory protein related lipid transfer domain-3 (STARD3) which is also known as Endosomal Metastatic Lymph Node protein 64 (MLN64), STARD3 N-terminal like (STARD3NL) which binds the membrane of the Late endosome and VAMP-Associated Protein (VAP) which binds the ER [1,19]. VAP, is an endoplasmic reticulum protein, plays a role as transitory contact between the ER membrane and the membrane of other organelles. STARD3 is specific for cholesterol and its N and C-terminals project into the cytosol [25].
Cholesterol movement from lysosome to peroxisome is accomplished by binding of cholesterol to NPC-1 N terminal protein which facilitates penetration of cholesterol into the lysosomal membrane. Free cholesterol is transferred from lysosome to peroxisome via membrane contacts between lysosome and peroxisome (Syt7 and PI(4,5)P2). [25,8]. Syt7 is a member of Synaptotagmin protein family, colocalized with the lysosome marker Lysosomal- Associated Membrane Protein 1 (LAMP1) and Peroxisome Marker PMP70, and plays important role in lysosomal exocytosis [25,26]. LAMP-1 and LAMP-2 are found in late endosomes and lysosomes as integral membrane glycoproteins. It has been shown that LAMP-2 has a main role in cholesterol transport across the late endosomal/ lysosomal compartments [25,26]. LAMP2A, mediates chaperonemediated autophagy by binding cytosolic protein substrates on the lysosomal membrane.
STARD3 (MLN64) transfers cholesterol from endosomes to mitochondria independently of NPC1 and it regulates late endosomal tethering [1,27].
D. Cellular removal of cholesterol
ATP Binding Cassette (ABC) proteins which comprise 48 types of transporters, twenty of them (e.g. ABCA1, ABCG1, ABCG4, ABCA5 and ABCA7) have a role in Reverse Cholesterol Transport (RCT) and phospholipid- and cholesterol efflux by transporting the compounds derived from lipid- or cholesterol metabolism across cellular membrane. ABCA1, ABCA7 and ABCG1 are major cholesterol cellular export proteins [26,27]. The role of ABCA1 is critical for the efflux of free cholesterol from peripheral cells. Cholesterol is transferred to lipid-poor Apo A-I particles, therefore this step is accepted as rate-limiting step in reverse cholesterol transport pathway. ABCA3 plays a role in translocation of phospholipids and cholesterol into lysosomal-like organelles (lamellar bodies) [1]. ABCG1 is involved in cholesterol efflux in macrophages and may regulate cellular lipid homeostasis in other cell types as well [28,29]. ABCA5 is proposed to play an important role in intracellular trafficking. But the substrates for ABCA5 and possible role of ABCA5 in human disease are not known clearly. Many diseases were defined related to the genetic mutations ABC transporters, one of them have high impact on cholesterol metabolism; Tangier Disease (TD) with mutation in ABCA1 [1,30] is an autosomal recessive disease caused by loss-of-function mutations in both ABCA1 alleles. Heterozygotes show a phenotype compatible with familial HDL deficiency (FHD), with low HDL-C linked to a reduction of about 50% in the ABCA1- mediated cell cholesterol efflux [31].
Conclusion
Although cholesterol metabolism inside of cell differs in different cell types and under different metabolic conditions, the main pathways and proteins are similar. Recently, it has been clearly shown that cholesterol was transferred between plasma membrane-late endosome-lysosome-peroxisome endoplasmic reticulum via carrier proteins or membrane contact site proteins inside cell. Various genetical disorders arise because of deficiency or defect of these proteins. So they are new therapeutic targets especially for diseases with hypercholesterolemia.
Acknowledgement
The authors grateful Gulce Selin Sozmen for drawing of Figure 1.
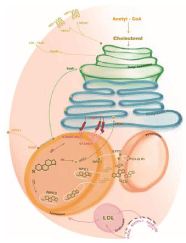
Figure 1: Cellular transport of cholesterol. (illustrated by G.Selin Sozmen).
References
- Van der Kant R, Zondervan I, Janssen L, Neefjes J. Cholesterol-binding molecules MLN64 and ORP1L mark distinct late endosomes with transporters ABCA3 and NPC1. J Lipid Res. 2013; 54: 2153-2165.
- Go G, Mani A. Low-Density Lipoprotein Receptor (LDLR) Family orchestrates cholesterol Homeostasis. Yale J Biol Med. 2012; 85: 19-28.
- Du X and Yang H. Endosomal cholesterol trafficking: protein factors at a glance. Acta Biochim Biophys Sin. 2013; 45: 11-17.
- Christoffersen M, Tybjærg-hansen A. Novel genes in LDL metabolism – a comprehensive overview. Curr Opin. 2015; 26: 179-187.
- Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin. 2013; 24: 332-328.
- Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest 2003; 111: 1795-1803.
- Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Review Cholesteryl ester storage disease : Review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol [Internet]. European Association for the Study of the Liver. 2013; 58: 1230-1243.
- Dubland JA, Francis GA. Lysosomal acid lipase: at the crossroads of normal and atherogenic cholesterol metabolism. Front Cell Dev Biol. 2015.
- Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, et al. Lysosomal acid lipase deficiency - An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014; 235: 21-30.
- Sigrid W. Fouchiera,b and Joep C. Defesche. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol 2013; 24: 332-338.
- Appelqvist H, Wa P. The lysosome : from waste bag to potential therapeutic target. J Mol Cell Biol. 2013; 5: 214-226.
- Yu XH, Jiang N, Yao PB, Zheng XL, Cayabyab FS, Tang CK. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin Chim Acta. 2014; 429: 69-75.
- Liu JP. New functions of cholesterol binding proteins. Mol Cell Endocrinol. 2009; 303: 1-6.
- Alobaidy H. Recent Advances in the Diagnosis and Treatment of Niemann- Pick Disease Type C in Children: A Guide to Early Diagnosis for the General Pediatrician. Int J Pediatr [Internet]. 2015; 2015: 1-10.
- Goldman SD, Krise JP. Niemann-Pick C1 functions independently of Niemann-Pick C2 in the initial stage of retrograde transport ofmembraneimpermeable lysosomal cargo. J Biol Chem . 2010; 285: 4983-94.
- Olkkonen VM, Li S. Oxysterol-binding proteins: sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Prog Lipid Res . 2013; 52: 529-538.
- Suchanek M, Hynynen R, Wohlfahrt G. The mammalian oxysterolbinding Protein-Related Proteins (ORPs) bind 25-hydroxycholesterol in an evolutionarily conserved pocket. Biochem J. 2007; 405: 473-480.
- Schultz ML, Tecedor L, Chang M, Davidson BL. Clarifying lysosomal storage diseases. J Cell Mol Med. 2011; 15: 280-95.
- Raiborg C, Wenzel EM, Stenmark H. ER–endosome contact sites: molecular compositions and functions. EMBO Journal. 2015.
- Raychaudhuri S, Im YJ, Hurley JH, Nonvesicular sterol movement from plasma membrane to ER requires oxysterol-binding protein-related proteins and phosphoinositides. J Cell Biol. 2006; 173: 107-119.
- Hamer I, Beersel G Van, Arnould T, Jadot M. Lipids and Lysosomes. Curr Drug Metab. 2012.
- Soccio RE, Breslow JL. Intracellular Cholesterol Transport. Arterioscler Thromb Vasc Biol. 2004; 24: 1150-1160.
- Urano Y,Watanabe H, Murphy SR,. Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc Natl Acad Sci U S A 2008; 105: 16513- 16518.
- Ganley IG, Pfeffer SR. Cholesterol accumulation sequesters Rab9 and disrupts late endosome function in NPC1-deficient cells. J Biol Chem. 2006; 281: 17890-17899.
- Chu BB, Ya-Cheng Liao, Bo-Liang Li, Bao-Liang Song. Cholesterol Transport through Lysosome-Peroxisome Membrane Contacts. Cell. 2015; 161: 291- 306.
- Jin Y, Strunk BS, Weisman LS, Michigan AA. Close encounters of the lysosome/peroxisome kind. Cell. 2015; 161:197-198.
- Charman M, Kennedy BE, Osborne N, Karten B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J Lipid Res. 2010; 51: 1023-1034.
- Vasiliou V, Vasiliou K, Nebert DW. Human ATP-binding cassette (ABC) transporter family. Hum Genomics. 2009; 3: 281-290.
- J. Štefková, R. Poledne, JA. Hubáček. ATP-Binding Cassette (ABC) Transporters in Human Metabolism and Diseases. Physiol. Res. 2004; 53: 235-243.
- Kuzaj P, Kuhn J, Dabisch-ruthe M, Faust I, Götting C, Knabbe C, et al. ABCC6- a new player in cellular cholesterol and lipoprotein metabolism ? Lipids Health Dis. 2014; 13: 118.
- Maranghia M, Truglioab G, Gallo A, Grieco E, Verrienti A, Montali A, et al. A novel splicing mutation in the ABCA1 gene, causing Tangier disease and familial HDL deficiency in a large family. Biochemical and Biophysical Research Communications . 2019; 508: 487-493.