
Case Report
J Endocr Disord. 2024; 9(1): 1050.
Facial Swelling in a 5-Year-Old Female
Taylor Dalsing, MD; Alison Coren, MD; Victor Kieu, MD; Bethany Auble, MD, Med*
Medical College of Wisconsin, Children’s Wisconsin, USA
*Corresponding author: Bethany Auble Department of Pediatrics, Medical College of Wisconsin, 9000 West Wisconsin Avenue, Milwaukee, Wisconsin 53226, USA. Tel: 414-266-6750 Email: bauble@mcw.edu
Received: February 22, 2024 Accepted: March 26, 2024 Published: April 02, 2024
Presentation
A previously healthy 5-year-old female presents to the emergency department with a chief concern of worsening facial swelling. She recently completed a course of Augmentin, prescribed at an urgent care for a presumed dental infection. Her weight is 26.9 kg (95th percentile), height is 116 cm (66th percentile), and BMI is 19.99 (97.5th percentile). Her initial vitals are significant for hypertension for age with a blood pressure of 122/79 mm Hg (99th percentile; 90th percentile 108/68) but are otherwise age appropriate. The emergency medicine team notes a protuberant abdomen on examination, raising concern for generalized edema. Laboratory evaluation is notable for normal electrolytes, BUN, creatinine, AST, ALT, albumin, urinalysis, and NT-proBNP. She also has a normal CXR and abdominal ultrasound. She is admitted for further evaluation.
On presentation to the acute care floor, our hospital medicine and endocrinology teams elicit additional history from the patient’s mother, which includes hyperphagia, weight gain, and a dramatic change in her appearance over the course of the previous 3 weeks (Figures 1A and 1B). There is a family history of early heart disease and stroke in her maternal grandfather (late 40s); there is no known family history of sudden death or early onset cancers/tumors. Repeat vital signs reveal persistent hypertension (128/85 mmHg). Physical exam performed by endocrinology notes moon facies and central adiposity, not edema as interpreted by previous providers. She has normal S1 and S2 heart sounds without murmur. Lungs are clear to auscultation. There is no hepatosplenomegaly, ascites, or masses. She does not have periorbital or lower extremity edema. She does not have flank tenderness. She does not have striae, acne, hirsutism, dorsocervical fat pad, extremity thinning, or other unusual skin changes. She was noted to have a baseline morning cortisol of 22.4 ug/dL (reference range 4.5-23.0 ug/dL). She received dexamethasone 1 mg at 11PM, and repeat morning cortisol at 7AM was 23.5 ug/dL (normal response <1.8 ug/dL). 24-hour free urine cortisol was 666.7 ug/24 hours (reference range 1-30).
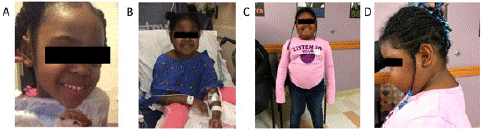
Figure 1: Photographs of patient taken over time. Photograph A was taken three weeks prior to initial presentation, B at initial presentation, C and D three months after initial presentation.
Discussion
Differential Diagnosis
Edema is characterized by fluid accumulation in various tissues. The differential includes, but is not limited to, heart and renal failure, cirrhosis, angioedema, protein malnutrition, protein losing enteropathy, and lymphedema. Cushing Syndrome (CS) can also cause edema through cross-reaction of cortisol with the mineralocorticoid receptor. The localization of the edema, including whether it is focal or generalized, can help point to an underlying diagnosis. It is also important to consider whether the patient truly has edema vs another process that makes tissue appear more full than normal, such as increased adiposity.
Actual Diagnosis
The patient presented with a chief concern of facial swelling. On examination, she was noted to have moon facies and central adiposity. Growth charts collected from her pediatrician’s office did not show significant growth deceleration; however, she did have rapid weight gain (Figure 2). The constellation of moon facies, central adiposity, rapid weight gain, and hypertension is highly suspicious for CS. CS describes a condition caused by prolonged exposure to supraphysiologic glucocorticoids. The patient’s diagnosis was cinched with documented hypercortisolism on two different tests. Although she did not have an appreciable dorsocervical fat pad or acanthosis nigricans (related to insulin resistance driven by hypercortisolism), these features became more apparent over time (Figure 1C and 1D). Growth deceleration is typically present in children with CS. It may have been absent in this patient because of the acuity in which her hypercortisolism developed.
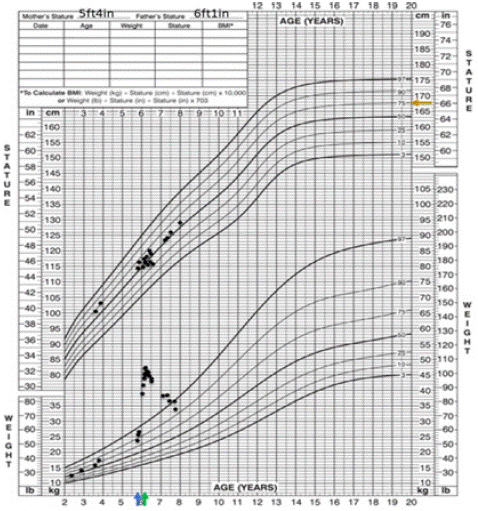
Figure 2: Growth chart. Blue arrow represents age at initial presentation. Green arrow represents age at bilateral adrenalectomy. Orange arrow represents mid-parental height.
The Condition
In a normally functioning Hypothalamic-Pituitary-Adrenal (HPA) axis, the hypothalamus secretes Corticotropin Releasing Hormone (CRH), which stimulates the anterior pituitary to release Adrenocorticotropic Hormone (ACTH), which then stimulates the adrenal gland to release cortisol. Cortisol acts by negative feedback to inhibit CRH release. The effects of cortisol are systemic. Its main function is to respond to stress, but it has a diurnal release that is important for physiologic functions, such as glucose homeostasis and blood pressure support. Cortisol levels are highest upon waking in the morning, followed by a steady decrease throughout the rest of the day and a nadir in the middle of the night.
Children with CS typically present with weight gain and decreased linear growth velocity [1]. Other signs and symptoms, not noted in our patient, include easy bruising, purple striae (more common in older children), hirsutism, amenorrhea, and mood changes [1]. Long-term sequelae of hypercortisolism include compromised adult height, impaired glucose metabolism, type 2 diabetes, hyperlipidemia, increased arterial rigidity, osteoporosis, neuropsychiatric disturbances, and decreased quality of life [1].
Exogenous administration of corticosteroids (e.g. prednisone) is the most common etiology. Endogenous etiologies of CS are much rarer, occurring with an incidence of 0.7-2.4 per million people per year [2]. CS results from dysregulation at any point along the HPA axis and includes both ACTH and non-ACTH mediated processes. Cushing disease is one etiology of CS and describes overproduction of ACTH by a pituitary adenoma. Other etiologies of CS include ectopic ACTH-secreting tumors. Adrenal overproduction of cortisol is seen in adrenocortical nodular disease.
Treatment/Management
Diagnosis begins with documenting hypercortisolism with 24-hour urine free cortisol, late-night salivary cortisol (=2 tests), or an overnight 1-mg dexamethasone suppression test. These tests are highly sensitive but also have a high false-positive rate [3]. Individuals with one positive test should be referred to an endocrinologist for ongoing testing, which generally involves performing one or two additional tests [3]. Individuals with a negative test but high pre-test probability should also be referred to an endocrinologist to exclude the possibility of cyclic hypercortisolism, which causes intermittent episodes of cortisol excess [3].
After documenting hypercortisolism, the next step involves distinguishing between ACTH-dependent (includes Cushing disease and ectopic ACTH production) and ACTH-independent Cushing syndrome. Individuals with ACTH-dependent hypercortisolism should have a pituitary imaging [4]. Inferior petrosal sinus sampling may be needed to detect microadenomas [4]. CT, MRI, or other nuclear imaging studies of the neck, chest, abdomen, and pelvis are used to detect ectopic sources of ACTH production [1]. Individuals with ACTH-independent CS should have adrenal imaging [4].
Transsphenoidal surgical resection of ACTH secreting pituitary adenomas is first line treatment of Cushing disease and success rate is 90% in specialized centers [1]. Risks involve hypopituitarism, bleeding, and infection. Pituitary radiotherapy is generally avoided in children due to risk for hypopituitarism and cognitive deterioration, but stereotactic radiotherapy may limit these adverse effects and is being studied in children [1]. Individuals with adrenal tumors are best treated with surgical resection, and those with micronodular or macronodular disease with bilateral total adrenalectomy. Medications that modulate ACTH release, inhibit adrenal steroidogenesis, and/or block the glucocorticoid receptor are second-line therapies [4]. These drugs are not currently FDA approved for the treatment of hypercortisolism and have significant side effects, so they are generally reserved for individuals who are not surgical candidates or have persistent disease after surgery [4].
Patient Course
Our patient was discharged as we awaited results from her first admission. ACTH was suppressed, and CT abdomen showed normal adrenal glands. There are several forms of adrenocortical disease that result in excessive cortisol production, not appreciable on imaging studies. These can be distinguished using the Liddle test, which measures urinary cortisol and 17-hydroxycorticosteroids at baseline and after giving low and high-dose dexamethasone. Among individuals with one form of adrenocortical disease, called Primary Pigmented Nodular Adrenocortical Disease (PPNAD), dexamethasone induces a paradoxical increase in cortisol [6], thought to occur through a glucocorticoid receptor-mediated effect on protein kinase A catalytic subunits [7]. In those with other types of adrenocortical disease, including macronodular adrenocortical disease and adrenal adenomas, this paradoxical increase is generally absent [6].
PPNAD is typically associated with Carney complex, which is a rare genetic disorder characterized by endocrine and non-endocrine tumors, including myxomas and schwannomas [5]. Most individuals with PPNAD have a genetic variant in the PRKAR1A gene, which codes for a regulatory subunit of protein kinase A. Our patient had a paradoxical response to dexamethasone and a variant in PRKAR1A, confirming the diagnosis of PPNAD in the setting of Carney complex.
She had a pre-operative evaluation with cardiology to assess for cardiac myxoma, which is a source of significant morbidity and mortality among individuals with Carney complex. Her cardiologist noted an echo-dense mass on echocardiography, raising concern for myxoma. She had a cardiac MRI, which has improved specificity, to further characterize the mass, and this study was normal, suggesting that the mass on echocardiography was likely an artifact. She was subsequently cleared for surgery. She underwent successful bilateral adrenalectomy. Pathology revealed a benign adrenal gland with cortical nodularity. She received peri-operative stress steroids and subsequently started physiologic hydrocortisone and fludrocortisone. Family also received education to administer stress dose hydrocortisone when sick and during stressful events. Hydrocortisone and fludrocortisone have been titrated based on symptoms, growth, electrolytes, and plasma renin levels [8].
She is followed by endocrinology, cardiology, and our cancer predisposition teams. She undergoes annual echocardiograms and close monitoring of her growth rate and pubertal staging. After puberty starts, she will undergo additional surveillance with thyroid and ovarian ultrasounds and whole-body MRI. At time of this writing, PPNAD is our patient’s only manifestation of Carney complex. Her appetite and weight are normalizing (Figure 2).
Lessons for Clinician
• It is important to maintain a wide differential diagnosis when patients present with non- specific chief complaints such as “swelling”.
• Comparing photographs over time may provide valuable insight into clinical characteristics for patients presenting with rapid weight gain of unknown etiology.
• Cushing syndrome must be considered in a child presenting with hypertension, linear growth deceleration, and rapid weight gain.
• The majority of pediatric Cushing syndrome is iatrogenic, secondary to steroid administration, but there are rare disorders of endogenous ACTH and/or cortisol production that led to Cushing syndrome.
• Cushing syndrome is typically caused by an adrenal source in children <7 years old and ACTH-producing tumors in older individuals.
Author Statements
Author Disclosure: Drs. Dalsing, Coren, Kieu, and Auble have disclosed no financial relationships relevant to this article. This commentary does not contain a discussion of an unapproved/investigative use of a commercial product/device.
References
- Lodish MB, Keil MF, Stratakis CA. Cushing Syndrome in Pediatrics. Endocrinol Metab Clin North Am. 2018; 47: 451-462.
- Sharma ST, Nieman LK, Feelders RA. Cushing’s syndrome: epidemiology and developments in disease management. Clin Epidemiol. 2015; 7: 281-93.
- Nieman LK, Biller BMK, Findling JW, Newell-Price J, Savage MO, Srewart PM, et al. The Diagnosis of Cushing’s Syndrome: An Endocrine Society Clinical Practices Guideline. JCEM. 2008; 93: 1526-1540.
- Nieman LK, Biller BMK, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline. JCEM. 2015; 100: 2807-2831.
- Kamilaris CD, Faucz FR, Voutetakis A, Stratakis CA. Carney Complex. Exp Clin Endocrinol Diabetes. 2019; 127: 156-164.
- Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999; 131: 585-91.
- Louiset E, Stratakis CA, Perraudin V, Griffin KJ, Libe R, Cabrol S, et al. The paradoxical increase in cortisol secretion induced by dexamethasone in primary pigmented nodular adrenocortical disease involves a glucocorticoid receptor-mediated effect of dexamethasone on protein kinase A catalytic subunits. JCEM. 2009; 94: 2406-2413.
- Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. JCEM. 2016; 101: 364-89.