
Abstract
In this work, we have performed theoretical investigations on the oxidation of trifluoromethyl methyl ether (CH3OCF3) molecule initiated by Cl atom using the Density Functional Theory (DFT) method. Here we have constructed an energy profile diagram for the three reaction channels viz. CH3OCF3 + Cl → C•H2OCF3 + HCl (R1), CH3OCF3 + Cl → CH3O• + CF3Cl (R2) and CH3OCF3 + Cl → CF3O• + CH3Cl (R3) along with the transition states at M06-2X/6-31+G (d,p) level of theory. Our energy profile result shows that R1 channel is found to be the lowest energy barrier reaction as compared to the other two channels. Thermochemical results also suggest that the product of R1 reaction is more stable than others. In addition to this, we have also determined heats of formation (ΔfH° 298) using isodesmic reactions method and Bond Dissociation Energy (BDE) of CH3OCF3 molecule. The value of ΔfH° 298 and BDE of the molecule are found to be good agreement with the value reported in the literature.
Keywords: HFE; DFT; Isodesmic; Heat of formation; BDE
Introduction
Hydrofluoroethers (HFEs) are a class of non-ozone depleting organic compounds. They serve as an alternative to Chlorofluorocarbons (CFCs), Hydrofluorocarbons (HFCs), and Hydrochlorofluorocarbons (HCFCs) for a wide range of commercial applications such as cleaning of electronic equipment, heat transfer fluid in refrigerators, lubricant deposition and foam blowing agents. Chlorine and bromine atoms are responsible for ozone depletion which remains absent in HFE [1,2]. The C-F bonds of HFEs absorb infrared radiation, thus acting as greenhouse gases [3]. Consequently, HFEs are considered to be the potential candidate for the global warming effect. In general, the HFEs having C-H bond, happens to get oxidized in the atmosphere by highly reactive radical species, such as OH radical and Cl atom. These reactions constitute the initial step of their degradation and thus results in short atmospheric lifetimes and Global Warming Potentials (GWPs) reducing their environmental impact. Also, the presence of an ether (-O-) linkage in HFEs enhances their reactivity in the troposphere, resulting in a shorter atmospheric lifetime which limits their accumulation in the atmosphere and decreases their potential impact as greenhouse gases [4]. Numerous studies on HFEs with OH radical has been explored both theoretically and experimentally [5-7]. The study of HFEs with Cl atom is also important in the marine regions, which can not be ignored. There are very limited studies are reported in the literature in this regard.
In the present work, we aim to address the degradation pathways of the trifluoromethyl methyl ether (CH3OCF3) with Cl atom using the quantum chemical method. The reaction between CH3OCF3 and Cl atom will proceed by H-atom abstraction as well as Cl atom addition and C-O bond breaking, leading to the formation of alkyl and alkoxy radicals. The following reactions are considered for the study and shown in Scheme 1.
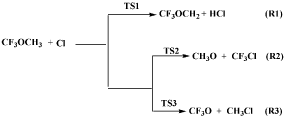
Scheme 1: Cl-atom initiated oxidation reactions of CH3OCF3 molecule.
Furthermore we will also determine the heat of formation (ΔfH° 298) and Bond Dissociation Energy (BDE) of CH3OCF3 molecule by using quantum chemical method. Here, we will determine heat of formation of CH3OCF3 molecule by using isodesmic reaction method [8] at the same level of theory. As the isodesmic reaction is a relatively inexpensive and accurate method to predict the enthalpy of formation because the conservation of the number of electron pairs in the reactants and products and the similarity of bond type cancel the possibilities of systematic errors in the calculations. Here, we predict the heat of formation for CH3OCF3 molecule, using the following isodesmic reactions:
CH3OCF3 + CH4 → CH3OCH3 + CHF3 (R4)
CH3OCF3 + CH3F → CH3OCH3 + CF4 (R5)
Bond dissociation energy (BDE) is an important parameter in understanding the progress of a chemical reaction. So, we are considered to obtain BDE of species using the following reaction
CH3OCF3 → C•H2OCF3 + H (R6)
Computational Details
Electronic structures and thermochemical calculations are performed using the Density Functional Theory (DFT) method as given in Gaussian 09 software package [9]. We have used M06- 2X functional [10] of DFT method along with 6-31+G(d) basis set to obtain optimized geometry of all species. It is important to note that 2X in M06-2X represented the double the amount of nonlocal exchange, and it is parametrized mainly for non-metals [11]. This functional has been chosen for the calculations as it served better results for thermochemistry, kinetics, and barrier height on related systems [1,12,13]. Further, we have performed vibrational frequencies calculations for all the species at the same level of theory. Frequency results stand hold of the stable minima that correspond to the real and positive values except for transition states. Transition states have been identified by only one imaginary frequency. Intrinsic Reaction Coordinate (IRC) calculations have been performed to testify the transition states smoothly connecting the reactant and product [14].
The Heat of reaction is calculated by using the expression:
ΔHr º= S Hº (Products) - S Hº (Reactants) (1)
Heat of reaction also expressed as:
ΔHrº= ΔHfº (Products) – ΔHfº (Reactants) (2)
Where H denotes enthalpy of the species and ΔHf º change of heat of formation of species
BDE of C-H bonds are calculated by using the given equation:
BDE (A-B) = H(A•) + H(B•) - HA-B) (3)
where H’s are the enthalpy of the corresponding species. A• and B• are the radical species formed as a result of the homolytic fission of the covalent bond.
Results and Discussion
Electonic Structures and Energy Profile Diagram
We have first optimized the geometry of CH3OCF3 molecule at M06-2X/6-31+G(d) level of theory and found that hydrogen atoms present in -CH3 sites are equivalent. Thus, we have considered only one H-abstraction reaction from –CH3 sites of CH3OCF3. In addition to this, we have also considered Cl-atom addition to CH3- and CF3- site of CH3OCF3 molecule and C-O bond cleavage reactions. All the possible reaction channels (R1-R3) are shown as Scheme 1.
Considering these reaction pathways, the geometries of all the species including Reaction Complexes (RC), Transition States (TSs), Product Complex (PC) and products are optimized at M06-2X/6- 31+G(d) level of theory and are presented in Figure 1 along with the bond lengths (in Å).
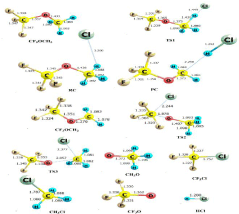
Figure 1: Optimized structures of all species of the reaction channels R1-R3
at M06-2X/6-31+G (d,p) level of theory.
We have observed from Figure 1 that in the case of H-abstraction by Cl-atom as given in R1, the C-H bond distances of TS1 increase from 1.088Å to 1.430Å with respect to equilibrium C-H bond of CH3OCF3. In R2, Cl-atom addition occurs at -CF3 site of CH3OCF3 molecule and simultaneously C-O bond breaks and thus leading to the formation of CH3O + CF3Cl via TS2. In TS2, the Cl is at a distance of 2.44 Å from C atom of CF3 site and at the same time C-O bond distance increases from 1.337 Å to 1.878 Å in TS2 with respect to CH3OCF3 molecule. In the same way, in R3 pathway, Cl-atom addition occurs at -CH3 site of CH3OCF3 molecule and breaking of the C-O bond is also observed simultaneously, leading to the formation of CF3O + CH3Cl via TS3. In TS3, the Cl-atom is at a distance of 2.38 Å from C atom of CH3 - site and at the same time C-O bond of TS3 increases with respect to CH3OCF3 from 1.43 Å to 2.37 Å.
Frequency calculations of all species have also been performed at M06-2X/6-31+G(d,p) level of theory. Vibrational frequency results signify the existence of stable minima, having real positive frequencies for all the species except the transition states. Transition states TS1, TS2 and TS3 are characterized by the occurrence of only one imaginary frequency observed at 760i, 543i and 724i cm-1. Visualization of the imaginary frequency has revealed a qualitative confirmation of the existence of transition states connecting reactants and products. Intrinsic reaction coordinate (IRC) calculations have been performed for each transition state at the same level of theory. Results show that each transition state connects the reactant and the product sides smoothly.
We have also explored the reaction pathways to determine the kinetically favorable one. Zero-point corrected total energy for all species and TSs along with the relative energy barrier are presented in Table 1.
![]()
Reaction Species
E0 (in Hartree)
R.E. (in kcal mol-1)
CH3OCF3 +Cl
-912.6752994
0
RC
-912.680852
-3.48
TS1
-912.6747171
0.37
TS2
-912.5540586
76.08
TS3
-912.5793497
60.21
PC
-912.678111
-1.76
CF3OC•H2 + HCl
-912.6748833
0.26
CH3O + CF3Cl
-912.6427967
20.40
C•F3O + CH3Cl
-912.6511897
15.12
Table 1: Zero-point corrected total energy (E0) for species and transition states along with the relative energy (R.E) at M06-2X/6-31+G (d,p) levels of theory.
A Potential Energy Diagram (PES) of the title reaction is constructed with the help of data reported in Table 1 and shown in Figure 2.
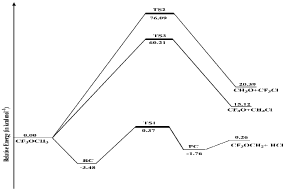
Figure 2: Potential energy diagram of CH3OCF3 + Cl reaction at M06-2X/6-
31+G (d,p) level of theory.
In the construction of the energy diagram, the ground state energies of the reactants are arbitrarily taken as zero. The calculated barrier heights for TS1, TS2 and TS3 are 0.37, 76.08 and 60.21 kcal mol-1, respectively. So R1 channel is faster than the other two reaction channels (R2 and R3). In other words, we can say that Cl-initiated H-abstraction reaction is more dominant channel than the other two channels. We further observed that the product obtain corresponding to R1 channel is more stable than the other two products. Whereas in case Cl-atom addition and simultaneous C-O dissociation reactions (R2 and R3), R3 pathway displayed more facile pathway to obtain CF3O and CH3Cl in comparison to R2 pathway.
Reaction energies (R1-R3), Heat of Formation and BDE of CH3OCF3 molecule
From frequency calculations, we have obtained absolute enthalpies and Gibbs free energies of all species (in Hartree). From these data, we have determined enthalpy and Free energy changes of the reaction channels (R1-R3) and are listed in Table 2.
![]()
Reaction Channels
ΔrH0
ΔrG0
CH3OCF3 + Cl→CF3OCH2+HCl (R1)
0.90
-1.45
CH3OCF3 + Cl→CH3O+CF3Cl (R2)
20.47
16.74
CH3OCF3 + Cl→CF3O+CH3Cl (R3)
15.26
32.64
Table 2: Thermochemical data (in kcal mol-1) for reaction channels (R1-R3) calculated at M06-2X/6-31+G(d,p) levels of theory.
Results show that the H-abstraction reaction i.e. R1 is slightly endothermic in nature with ΔrH0 value of 0.90 kcal mol-1 but thermodynamically facile with ΔrG0 value of -1.45 kcal mol-1. On the other hand, Cl-atom addition and simultaneous C-O dissociation reactions (R2 &R3) are highly endothermic (ΔrH0 › 0) as well as thermodynamically not feasible (ΔrG0 › 0). These results are in accordance with the observed relative energy calculations.
In order to the find heat of formation and bond dissociation energy of CH3OCF3 molecule, geometry optimizations of all species present in the reactions R4-R6 are performed. From this, we have obtained a global optimized structure corresponding to minimum energy and further performed frequency calculations of species to get thermochemical data of all the species present in reactions. The absolute enthalpies values of all the species are given in Table 3.
![]()
Species
Enthalpy (H)
CH3OCF3
-452.568
CH4
-40.4389
CH3OCH3
-154.869
CHF3
-338.106
CH3F
-139.644
CF4
-437.339
C••H2OCF3
-451.913
H
-0.494
Table 3: Value of enthalpies (in Hartree) for all species present in R4-R6 at M06- 2X/6-31+G(d,p) level of theory.
The reaction enthalpies of the reaction (R4 and R5) and heat of formation of CH3OCF3 molecule are calculated by using Equation.1 and Equation.2 (described in the compuational details section) for isodesmic reactions R4 and R5. The experimental values of heat of formation (ΔfH° 298) for all species involved in the above reaction have been taken from data available at NIST [12]. The value of heat of formation is given in Table 4.
![]()
Molecule
Reactions
Heat of formation (ΔfH0298)
Literature values
CH3OCF3
R4
-212.70
R5
-215.77
Average
-212.74
(-212.7 ± 2.3) [15]
BDE
CH3OCF3
R6
100.73
(102 ± 1) [16]
Table 4: Heat of formation and Bond Dissociation Energy (BDE) of CH3OCF3 is calculated at M06-2X/6-31+G(d,p) level of theory. All values in kcal mol-1.
Conclusions
We have systematically investigated the reaction mechanism and thermochemistry of trifluoromethyl methyether CH3OCF3 (HFE- 143a) with Cl atom as well as Cl-atom addition and simultaneous C-O dissociation reaction with CH3OCF3 molecule at M06-2X/6- 31+G (d, p) level of theory. From the energy profile diagram we found that hydrogen atom abstraction by Cl atom follows the lowest barrier path with an activation energy of 0.37 kcal mol-1 than the other two channels. The thermochemical analysis reveals that R1 channel is slightly endothermic (ΔrH0 › 0) and thermodynamically facile (ΔrG0 ‹ 0), while R2 and R3 displayed highly endothermic nature (ΔrH0 › 0) and thermodynamically not feasible (ΔrG0 › 0). In addition to this, we have also obtained heat of formation of the CH3OCF3 molecule using isodesmic reaction schemes as well as bond dissociation energy of the molecule. These values are found to be good agreement of experimentally reported values. The presented work would be very much helpful to understand oxidation pathways and thermochemistry of other such species. This study would also be helpful to determine kinetics, lifetime and global warming potential of HFEs.
Acknowledgment
Dr. S. Paul is thankful to the University Grant Commission (UGC), New Delhi for providing financial support from Dr. D. S. Kothari Post- Doctoral Fellowship (Award letter no: F.4-2/2006(BSR)/ CH/16-17/0152). Dr. N.K. Gour and Mr. S.D. Baruah are thankful to DST, New Delhi for providing DST Nanomission project (SR/NM/ NS-1147/2016(G)) and DST INSPIRE fellowship [No: DST/INSPIRE Fellowship/(IF160658)], respectively.
References
- Hammit JK, Camm F, Connell PS, Mooz WE, Wolf KA, Wuebbles DJ, et al. Future emission scenarios for chemicals that may deplete stratospheric ozone. Nature. 1987; 330: 711-716.
- Gour NK, Deka RC, Singh HJ and Mishra BK. Theoretical studies on kinetics, mechanism and thermochemistry of gas-phase reactions of CF3CHFCF2OCF3 with OH radicals and Cl atoms and fate of alkoxy radical at 298 K. Journal of Fluorine Chemistry. 2014; 166: 110-116.
- Bera PP, Francisco JS, Lee TJ. Design strategies to minimize the radiative efficiency of global warming molecules. Proceedings of the National Academy of Sciences. 2010; 107: 9049-9054.
- Ponnusamy S, Sandhiya L, Senthilkumar K. Atmospheric Oxidation Mechanism and Kinetics of Hydrofluoroethers, CH3OCF3, CH3OCHF2, and CHF2OCH2CF3, by OH Radical: A Theoretical Study. The Journal of Physical Chemistry A. 2018; 122: 4972-4982.
- Wu JY, Liu JY, Li ZS, Sun CC. Theoretical study of the reactions of CF3OCHF2 with the hydroxyl radical and the chlorine atom. ChemPhysChem. 2004; 5: 1336-1344.
- Sulbaek Andersen MP, Nielsen OJ, Wallington TJ, Hurley MD, DeMore WB. Atmospheric chemistry of CF3OCF2CF2H and CF3OC(CF3)2H: reaction with Cl atoms and OH radicals, degradation mechanism, global warming potentials, and empirical relationship between k (OH) and k (Cl) for organic compounds. The Journal of Physical Chemistry A. 2005; 109: 3926-3934.
- Fontana G, Causà M, Gianotti V, Marchionni G. Computational studies of the reaction of the Hydroxyl radical with Hydrofluorocarbons (HFCs) and Hydrofluoroethers (HFEs). Journal of Fluorine Chemistry. 2001; 109: 113- 121.
- Ponomarev DA, Takhistov VV. What are isodesmic reactions?. Journal of chemical education. 1997; 74: 201.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. Gaussian 09, Rev D. 01, Wallingford, CT, USA. 2009.
- Zhao Y, Truhlar DG. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 2008; 120: 215-241.
- Gour NK, Mishra BK, Hussain I, Deka RC. Theoretical investigation on the kinetics and thermochemisty of H-atom abstraction reactions of 2-chloroethyl methyl ether (CH3OCH2CH2Cl) with OH radical at 298 K. Structural Chemistry. 2016; 27: 1491-1499.
- Srinivasulu G, Rajakumar B. Theoretical Investigations on the Kinetics of H-abstraction Reactions from CF3CH(OH)CF3 by OH Radicals. The Journal of Physical Chemistry A. 2013; 117: 4534-4544.
- Chase MW, Davies CA, Downey JR, Frurip DJ, McDonald RA. JANAF thermochemical tables: II: Cr-Zr. Journal of Physical and Chemical Reference Data. 1985; 14: 927-1856.
- Gonzalez C, Schlegel HB. An improved algorithm for reaction path following. The Journal of Chemical Physics. 1989; 90: 2154-2161.
- Good DA, Francisco JS. Bond dissociation energies and heats of formation for fluorinated ethers: E143A (CH3OCF3), E134 (CHF2OCHF2), and E125 (CF3OCHF2). The Journal of Physical Chemistry A. 1998; 102: 7143-7148.
- Hsu KJ, DeMore WB. Temperature-dependent rate constants and substituent effects for the reactions of hydroxyl radicals with three partially fluorinated ethers. The Journal of Physical Chemistry. 1995; 28: 11141-11146.