
Abstract
The objective of this study is to determine the residues of Organochlorine Pesticides (OCPs) residues in water of both Edko lake and fish farm in Abu Hummus region at El-Behira Governorate, Egypt. The analytical method included Solid Phase Extraction (SPE), for extraction and clean-up, followed by determination of OCP residues using GC-ECD and GC-ITD. A total of 18 OCPs were analyzed, only five compounds, heptachlorepoxide, p,p-DDE, dieldrin, p,p-DDD and endrin ketone were detected in the water of both Edko lake and fish farm. Their concentrations were 0.2309±0.0404, 1.3524±0.0311, 0.4104±0.0210, 1.2622±0.0218, and 0.1087±0.0212 µg/L, respectively, in water of Edko Lake and 0.3269±0.0221, 2.3479±0.0156, 2.2501±0.1553, 2.3466±0.0537, and 0.3092±0.0156 µg/L, respectively, in water of fish farm. Thus, the concentrations of OCP residues were higher in the water of fish farm than those in the water of Edko lake.
Keywords: Organochlorine pesticides (OCPs); SPE; Edko lake; GC-ECD; GC-ITD
Introduction
Water pollution with pesticides has been very serious problem to aquatic ecosystems because of the damage to aquatic species [1]. The Organochlorine Pesticides (OCPs) were reported as carcinogenic, mutagenic, endocrine-disrupting, persistent, and bio-accumulative chemicals [2]. These pesticides cause significant environmental and health problems due to their bioaccumulation in food chain. Once in aquatic environment, pesticides are absorbed by aquatic organisms and concentrated in trophic food chain thus endangering those organisms [3-6].
More than four decades ago, OCP pesticides were banned worldwide and by the Egyptian authorities because of their longpersistence, lipid solubility, and toxicity to humans and animals but are still in use in some developing countries, even though many new broad-spectrum pesticides have been developed in recent years [7].
Therefore, scientists applied several analytical methods in the extraction of wide range of OC pesticide residues from water. The analysis of pesticide residues depends on the use of sensitive and fast tools. Therefore, gas chromatography coupled with various detectors was proven suitable. The GC-MS has become a standard laboratory instrument that provide qualitative and quantitative results for analytes in a single injection. The most common MS technique uses a very rugged and practical quadrupole design, along with ionization (Electron Impact (EI)) [8]. More importantly, the validation of the analytical method is a requirement in the practice of pesticide residue analysis. Including the specificity, accuracy and precision, at relevant analyte concentrations and in appropriate matrices [9]. Therefore, the objective of this study was to compare the levels of organochlorine pesticides in water of both Edko lake and fish farm at El-Behira Governorate, Egypt.
Materials and Methods
Pesticides and reagents
A mixture of certified standard pesticides (2000±0.5 µg/ml) of a-HCH (99.7%), γ-HCH (99.9%), ß-HCH (98.9%), Δ-HCH (99.5%), heptachlor (99.9%), aldrin (98.9%), heptachlorepoxide (99.9%), endosulfan I (99.9%), endosulfan II (99.9%), endosulfan sulfate (99.4%), p,p-DDE (99.2%), p,p-DDT (98.9%), p,p-DDD (96.1%), dieldrin (99.2%), endrin (96.9%), endrin aldehyde (98.4%), endrin ketone (99.5%), and methoxychlor (99.9%) were obtained from SUPLECO company (Bellefonte, PA, USA). Standard solutions of 500ng of each pesticide/ml were prepared in methanol of and stored at -4oC. Working standard solutions were obtained by diluting the stock solutions with methanol.
Solid-phase extraction disk (Sep-Pak Plus) packed with 500mg of C18, Primary Secondary Amine (PSA), sodium sulfate, and magnesium sulfate were obtained from Milford, MA, USA. The solvents ethyl acetate, methylene chloride, methanol, and acetonitrile were of analytical grade and purchased from Merck, Darmstadt, Germany through reputed local suppliers.
Study areas and samples collection
Surface water samples were collected from two major sources at El-Behira Governorate, Egypt: Edko Lake and fish farm at Abu Hummus region (Map 1). The laboratory tap water was sampled as blank. Approximately, 9 samples (1 L each) of surface water were collected in glass bottles fitted with Teflon-lined caps from each location. The water samples were transferred immediately to the laboratory for extraction and analysis.
Map 1: Sites of collected water samples: (A) Edko Lake (river estuary) and
(B) Abu Hummus fish farm. Source: maps.google.com.
Extraction and clean-up of pesticide residues from water samples
Solid Phase Extraction (SPE) technique using Empore disc technique according to EPA method #3535 [10] with minor modifications was used to extract OCP residues from water samples. The disc was inserted into a filter apparatus and washed with 5 ml of a 1:1 mixture of ethyl acetate and methylene chloride. Then the disc was pre-wetted with 5ml methanol and rinsed with 5ml of diionized water. About 5ml of methanol were mixed well with each liter of water samples then passed through the reservoir under full vacuum. Extraction of samples were done via draining water from the sample container through discs. Then discs were rinsed with ethyl acetate the with methylene chloride. The combined eluates were dried through tubes of 5-7 g of anhydrous sodium sulfate. Extracts were concentrated to 0.5 - 1 ml gently over a water bath under a gentle stream of nitrogen.
Determination of pesticide residues in water
Extracts of water (1-2 µl) were analyzed for 18 OCPs utilizing Varian GC-ECD system (model 3400, Walnut, Greek, CA, USA). The GC was equipped with a Varian 8200 autosampler in splitless mode and maintained at 25oC. Chromatographic separation was achieved using HP-608 fused silica capillary column (30 m X 0.53 mm i.d. 0.5 µm film thickness). Helium was used as the carrier gas and nitrogen as the makeup gas. Separation conditions were as the following: the initial column temperature was set at 80° C for 6 min, increased to 215o C (for 1 min) at a rate of 15oC/min, then to 230o C at a rate of 5oC/min, and finally to 290oC (for 2 min) at 5oC/min.
Identification and confirmation for presence the targeted compounds in the investigated water samples were accomplished using a bench top Gas Chromatography-Ion Trap Detector, (GCITD), which consisted of a Varian 3800 series Gas Chromatograph interfaced to a Saturn 2000 equipped with a split/splitless part. The system was operated in splitless mode (purge time set at 1 min) and maintained at 250o C. All chromatographic separation was achieved using an HP-5MS capillary column (30m x 250µm id and 0.25µm film thickness). The carrier gas was helium at a constant flow rate of 1.1ml/min. The temperature at the column was initially set at 85oC for 0.3min, increased to 150oC (for 4 min) at a rate of 30oC/min, then to 185oC at a rate of 2oC/min and finally to 290oC (for 5 min) at a rate of 4oC/min. The ITD was operated in Electron Impact ionization mode (EI) at 70eV and temperature at 220o C. The EI spectra were monitored by scanning the ions within the range of 50-500 amu.
The targeted OC compounds were identified by their full scan mass spectra and retention time using the total ion current as a monitor to give the Total Ion Chromatogram (TIC). The use of the full scan mode allows comparing the spectrum obtained for interested compounds with the EI-MS library. In addition, Selective Ion Monitoring (SIM) mode was used for identification (mass spectra in elution time window) and searched for 3 selective specific ions for the compound of interest (Table 1).
![]()
OCPs
Ions, m/z
OCPs
Ions, m/z
a-HCH
183, 219, 111
Dieldrin
79, 263, 277
γ-HCH
181, 109, 219
Endrin
81, 263, 67
ß-HCH
181, 109, 219
p,p-DDD
235, 165, 199
Heptachlor
100, 272, 237
Endosulfan π
195, 207, 241
Δ-HCH
109, 183, 219
p,p-DDT
235, 199, 165
Aldrin
66, 79, 263
Endrin aldehyde
67, 345, 250
Heptachlorepoxide
81, 253, 263
Endosulfan sulfate
387, 272, 237
Endosulfan I
241, 195, 339
Methoxychlor
227, 308, 238
p,p-DDE
246, 176, 318
Endrin Ketone
67, 139, 317
Table 1: Major ions of the 18 OCPs using GC-ITD, which were used for the confirmation of these compounds in the investigated water samples.
Linearity and calibration of GC-ECD response
For quantification of OCPs, about 1-2 µL from each sample was injected into the GC-ECD that was operated under conditions described before. The concentration of each OCPs compound was determined from a calibration curve. The calibration curves (5 points) were constructed using OCPs-fortified ultra-pure water at concentration levels from 0.01 to 10 µg/L of each compound. The fortified blank-water samples were extracted and analyzed using the analytical procedure as described before. The integrated peak areas were plotted versus the concentration of the fortified samples. To check the linearity of the calibration graphs, the correlation of coefficient R2 for each compound was calculated.
Limits of Detection (LOD) and Limit of Quantification (LOQ)
The LOD and LOQ were determined according to PAM [11] and EPA [12]. The LOD was determined using the fortified-water samples with OCPs standard mixtures. It was calculated as the lowest concentration of the OCPs, which provides a chromatographic peak height 3 times the average baseline noise (at the same retention time). The limit of quantification was determined as the value of 10 times the baseline noise in GC-ECD chromatogram of the blank sample.
Method precision
Repeatability (intra-day assay precision) and intermediate precision (inter-day assay precision) were determined. The intra-day and inter-day precision of the method were determined by repeating analysis of 5 fortified laboratory blank water (0.1µg/L) on the same day and on 5 consecutive days, respectively. The average percentage of recovery for each compound and the relative standard deviation (%RSDs) were calculated.
Selectivity
The selectivity of the proposed analytical procedure was determined by the analysis of a solution of reference standards mixture in comparison with fortified and non-fortified blank water using GCECD. Additionally, using GC-ITD-SIM technique provides a very selective detection for analytes in investigated samples corresponding to the ions related to each analyte as mentioned in Table 1.
Extraction efficiency (Recovery tests)
The efficiency of SPE-Empore–Disc C18 approach as extraction tool of the 18 OCPs pesticide residues from water samples was assessed. The average percentage of recoveries (%Rec) and percent relative standard deviation (%RSD) were calculated for fortified blank samples. For that purpose, tap water (blank) was fortified with the mixture of 18 OCPs to reach a final concentration of 0.1 and 1 µg/l. Samples were extracted and analyzed as previously mentioned. The average percentages of recoveries (%Rec) and percentages of relative standard deviation (%RSD) for recoveries were calculated. All data of residue analysis were corrected according to recovery percentage values.
Results and Discussions
The analytical method was developed and validated for the determination of 18 organochlorine pesticides residues in water samples. Solid Phase Extraction (SPE) technique using Empore disc technique according to EPA 3535 (Tomkins et al., 1992) with minor modifications was used to extract OCP residues from water samples. Qualitative and quantitative analysis for the 18 OCPs residues in water samples were done using GC-ECD. Furthermore, GC-MS (GCITD) was used for confirmation and identification of the presence of targeted OCP residues. The GC-ECD and GC-MS conditions were optimized and the performance of proposed assay was done by the estimation of accuracy (% recovery), linearity range, Limit of Detection (LOD), limit of Quantification (LOQ), and selectivity [13,14].
GC-ECD and GC-MS conditions and optimization
Results in Table 2 summarize the chromatographic data, average Retention times (tR), Resolution (R), Tailing factor (T), and %RSD of tR. Data showed that all OCPs were resolved from each other at the baseline with resolution values ranged between 1.5 and 2.5. The separated peaks on GC-ECD chromatogram have a symmetrical shape with Tailing factor (T) ranged between 1.01 and 1.04. The % RSD values for the Rt were less than 1.5, indicating the stability of the chromatographic system. The chromatographic data were in accordance with USP requirements, which ensure the suitability and effectiveness of chromatographic system for qualitative and quantitative analyses of OCPs residues.
![]()
OCPs
tRa
Rb
Tc
a-HCH
13.31 ± 1.1
2.3
1.01 ± 0.025
γ-HCH
14.30 ± 1.2
2.1
1.02 ± 0.021
�-HCH
15.20 ± 0.9
2.2
1.01 ± 0.025
Heptachlor
15.37 ± 1.3
2.1
1.02 ± 0.036
Δ-HCH
16.40 ± 1.4
2.2
1.01 ± 0.015
Aldrin
17.47 ± 1.1
2.2
1.03 ± 0.025
Heptachlorepoxide
18.61 ± 1.2
2.5
1.02 ± 0.010
Endosulfan I
19.10 ± 1.1
2.4
1.02 ± 0.012
p,p-DDE
19.80 ± 0.8
2.3
1.04 ± 0.015
Dieldrin
20.80 ± 1.1
1.5
1.02 ± 0.015
Endrin
20.70 ± 1.1
1.5
1.03 ± 0.025
p,p- DDD
21.47 ± 1.3
1.6
1.04 ± 0.015
Endosulfan π
21.85 ± 1.1
1.8
1.01 ± 0.021
p,p-DDT
22.32 ± 1.2
1.5
1.01 ± 0.015
Endrin aldehyde
22.72 ± 1.1
1.5
1.02 ± 0.012
Endosulfan sulfate
23.08 ± 1.5
1.5
1.02 ± 0.015
Methoxychlor
24.50 ± 1.2
2.5
1.01 ± 0.021
Endrin Ketone
25.22 ± 1.1
2.3
1.02 ± 0.026
aReproducibility of tR for each analyte was evaluated during 2 months with minimum of 10 injections of reference standard mixture solution and the %RSDs were determined; bresolution was determined according to USP (2010) and the acceptable limit, R not less than 1.5; ctailing factor (T) was determined according to USP (2010) and the acceptable limit, T = 0.9 - 1.1.
Table 2: Chromatographic data obtained from injection of multi-standards of 18 OCPs into GC-ECD operated under optimized conditions.
From MS spectrum results, 3 ions for each pesticide were compared with the corresponding 3 ions that resulted from the analysis of reference standard mixture using GC- MS (GC-ITD) (Table 1). Results revealed that GC-ECD and GC-ITD were simple and rapid analytical tool (chromatographic run time about 30 min) for separation, identifications/confirmation, and quantifications of the 18 OCPs under investigation in water samples. The combination of chromatographic retention data and MS data might be used for positive identification of compounds through the use of analytical reference standards.
Determination of the accuracy of the analytical methods
The accuracy of the employed analytical method was determined by calculating average percentages of recoveries and percentages of relative standard deviation (%RSD) for all OCPs of fortified blank water samples. Average recoveries percentages were ranged from 92.1±7.1 to 101.2±5.1% and 92.6±6.1 to 102.3±4.0% at low and high levels of water fortifications, respectively (Table 3). These results were in agreement with those of Abbassy et al. (2010) who reported that the average recovery percentages ranged from 85.5±8.1 to 98.1±6.5% after the SPE of OCPs in water samples.
![]()
OCPs
(Rec%)±RSD
0.1µg/L
(Rec%)±RSD
1µg/L
a-HCH
99.1±6.1
99.2±6.2
γ-HCH
93.2±7.1
92.6±6.1
β-HCH
98.1±6.2
99.1±4.1
Heptachlor
93.7±7.1
92.8±6.2
Δ-HCH
101.2±5.1
100.2±3.2
Aldrin
95.4±6.2
94.3±5.2
Heptachlorepoxide
94.2±7.1
93.8±4.2
Endosulfan I
99.2±5.2
98.1±4.5
p,p-DDE
97.1±6.2
98.1±3.2
Dieldrin
98.2±7.2
99.1±1.2
Endrin
99.5±6.1
95.1±1.4
p,p-DDD
94.6±6.2
95.6±1.5
Endosulfan π
100.2±6.1
101.2±5.1
p,p-DDT
92.1±7.1
102.3±4.0
Endrin aldehyde
98.1±3.2
99.1±3.1
Table 3: Average recovery percentages (Rec%) and relative standard deviation (RSD%) for the 18 OCPs extracted from spiked water samples using SPEEmpore Disc technique.
Quantification, linearity and linearity range, and calibration of GC-ECD response
For quantification of OCPs in extracts of water samples, about 1-2 µl was injected into the GC-ECD. The concentration of each compound, determined from a 5 points calibration curve, was ranged from 0.01 to 10 µg/L. In order to check the linearity of the calibration graphs, correlation coefficient (R2) for each compound was calculated. The calibration data showed good linearity for the response of ECD detector for all tested analytes at concentration within the tested range and linear correlation coefficient higher than 0.99997.
Limit of Detection (LOD) and Limit of Quantification (LOQ)
The LOD was determined using the fortified laboratory blank water samples with the 18 OCPs standards mixture. The LODs were calculated as the lowest concentration of OCPs which provides a chromatographic peak height of 3 times the average baseline noise (at the same retention time). The limit of quantification (LOQ) was determined as the value of 10 times the baseline noise in GC-ECD chromatogram. The LOD and LOQ for analysis of 18 OCPs in water samples using the proposed method were found to be in the range 0.01-0.02 µg/l and 0.035- 0.075 µg/l, respectively.
Assessment the precision
Precision, repeatability (intra-day-assay precision), and intermediate precision (inter-day assay) were determined. The intraday and inter-day precision were determined by repeating analysis of 5 fortified blank samples with 0.1 µg/L on the same day and 5 on consecutive days, respectively (Table 4). Data showed that the developed method was precise as the RSD% values were less than 20% as illustrated by IEH [13] guidelines. This demonstrates the precision of the method and its effectiveness for quantitative purposes.
![]()
OCPs
Inter-day precision
%Rec.±RSD
Intra-day precision
%Rec.±RSD
a-HCH
98.1±4.1
99.1±5.1
γ-HCH
90.2±5.1
91.2±7.2
β-HCH
93.2±6.1
97.1±7.2
Heptachlor
98.2±5.1
93.2±7.1
Δ-HCH
94.8±3.0
99.2±6.1
Aldrin
97.9±3.9
94.9±5.1
Heptachlorepoxide
94.9±4.1
93.9±6.3
Endosulfan I
97.1±5.1
98.1±4.5
p,p-DDE
95.2±3.2
96.2±6.2
Dieldrin
96.1±2.2
96.8±5.1
Endrin
97.1±6.5
98.2±7.3
p,p-DDD
92.1±4.1
93.3±6.1
Endosulfan π
97.1±6.5
98.2±6.7
p,p-DDT
91.1±4.1
92.3±7.1
Endrin aldehyde
95.2±5.1
96.2±6.1
Endosulfan sulfate
91.2±4.1
90.8±5.6
Methoxychlor
92.3±5.1
91.2±4.5
Endrin Ketone
93.2±3.4
92.3±4.2
Table 4: Inter-day and intra-day precision data obtained from the analysis of multi-standards of OCPs in the fortified blank water (0.1μg/L) samples, extracted using SPE-Empore Disk technique, and analyzed using GC-ECD.
Selectivity of the analytical method
The selectivity of the proposed analytical procedures was determined by analysis of mixture solution of standard materials and compared with fortified and non-fortified blank samples. Results in Figure 1 showed no interfering peaks from the endogenous matrix. The resolution values under the selected operation conditions of GC-ECD and GC-MS were adequately resolved from each other. Additionally, the SIM technique provided very selective detection method for analytes (Table 1). All these validated data of the developed method for analysis of 18 OCPs in extracts of water were deemed acceptable according to IEH [13]. The method was precise, selective, accurate, and sensitive for the determination of 18 OCPs residues in extracts of water samples.
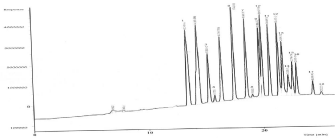
Figure 1: GC-ECD chromatogram of fortified laboratory water sample with
0.1µg/l of multi-standard of 18 OCPs that were extracted using SPE-Empore
Disc technique: 1) a–HCH; 2) γ-HCH; 3) ß-HCH; 4) Heptachlor; 5) Δ-HCH; 6)
Aldrin; 7) Heptachlorepoxide; 8) Endosulfan I; 9) p,p-DDE; 10) Dieldrin; 11)
Endrin; 12) p,p-DDD; 13) Endosulfan π; 14) p,p-DDT; 15) Endrin aldehyde;
16) Endosulfan sulfate; 17) Methoxychlor; and 18) Endrin Ketone.
Residue levels of OCPs in water samples collected from investigated areas
The developed and validated analytical method was applied for the analysis of residues of 18 OCPs in water samples from both Edko lake and Abu hummus fish farm. The data in Table 5 showed that only five OCP residues were detected in water of both Edko lake and fish farm. The average concentrations of heptachlorepoxide, p,p- DDE, dieldrin, p,p-DDD, and endrin ketone were 0.2309±0.0404, 1.3524±0.0311, 0.4104±0.0210, 1.2622±0.0218, and 0.1087±0.0212 µg/l, respectively, in Edko Lake and 0.3269±0.0221, 2.3479±0.0156, 2.2501±0.1553, 2.3466±0.0537, and 0.3092±0.0156 µg/l, respectively, in fish farm. The presence of OCP residues in surface water might be attributed to extensive use of parent pesticides in Egypt during the period from 1950 to 1970 in agricultural practices and slow degradation and high accumulation patterns in soils of agricultural fields [15]. On the other hand, a-HCH, ß-HCH, γ-HCH, heptachlor, Δ-HCH, aldrin, endosulfan I, endrin, endrin aldehyde, endosulfan II, endosulfan sulfate, p,p-DDT, and methoxychlor were not detected in water samples.
![]()
OCPs
tR
Residue (µg/L)±SD
Edko lake
Fish farm
a-HCH
13.31
ND
ND
γ-HCH
14.30
ND
ND
�-HCH
15.20
ND
ND
Heptachlor
15.37
ND
ND
Δ-HCH
16.40
ND
ND
Aldrin
17.47
ND
ND
Heptachlorepoxide
18.61
0.2309±0.0404
0.3269±0.0221
Endosulfan I
19.10
ND
ND
p,p-DDE
19.80
1.3524±0.0311
2.3479±0.0156
Dieldrin
20.80
0.4104±0.0210
2.2501±0.1553
Endrin
20.70
ND
ND
p,p-DDD
21.47
1.2622±0.0218
2.3466±0.0537
Endosulfan π
21.85
ND
ND
p,p-DDT
22.32
ND
ND
Endrin aldehyde
22.72
ND
ND
Endosulfan sulfate
23.08
ND
ND
Methoxychlor
24.50
ND
ND
Endrin Ketone
25.22
0.1087±0.0212
0.3092±0.0156
Table 5: Levels of the organochlorine residues (μg/L ± SD) in water samples collected from investigated areas in El-Beheira Governorate.
Despite the absence of DDT, heptachlor, aldrin, and endrin insecticides, degradation products such as DDE, DDD, heptachlor epoxide, dieldrin, and endrin ketone were dominant in water samples. This may be due to the different mechanisms of degradation such as microbial effect and oxidation [15], photoionization, electron transfer [16,17] and aerobic biodegradation, hydrolysis and photodegradation in surface water [18].
In general, current results revealed that detected OCP residues in water of the fish farm were higher than those in the water of Edko lake. This means that fish farm water was more contaminated than Edko lake water. The concentrations of these pesticide residues in water of both sources were exceeding the permissible limits proposed by WHO (1995) and Egyptian Ministry of Health Regulation report [19] for the drinking water. Fortunately, Egyptian people did not use water of both Edko lake and fish farm in drinking. The European Economic Community [20] and [21] have strict legislation concerning the occurrence of pesticide chemicals in water intended for human consumption, the Maximum Residual Level (MRL) of a pesticide chemical must be below 100ng/L and sum of all pesticides not exceed 0.5µg/L in surface water. Concentrations of pesticides greater than MRLs represent hazardous effects on human and environment health. Organochlorine pesticide residues or their degradation products were detected, for example endrin was detected as endrin aldehyde and endrin ketone, endosulfan was detected as endosulfan sulfate, DDT as DDD and DDE, heptachlor as heptachlor epoxide [22].
The presence of pesticides and their transformation products in water is in trace amounts (usually below the ppb levels). Therefore, extraction and sample preparation are critical steps for accountable result. The sample preparation employed herein was Solid Phase Extraction (SPE) and it was reported sensitive and reliable based on method validation parameters that were monitored. Our results were in agreement with several authors [7,23-25].
Conclusions
Monitoring environmental pollutants especially pesticide residues is very critical. The analysis method must be specific and accurate for such determination. Therefore, herein we validated an analytical method for the determination of OCPs and their degradation products in surface water resources in Egypt. The analytical method showed recovery percentages of more than 92% and accuracy that met the acceptable levels of ICH (2005) with recoveries from 70 to 130 % and RSD below 20 %. Moreover, the LOD and LOQ of the analytical method was from 0.01 to 0.02 µg/L. Application of the developed analysis method in detection of OCP residues in water samples of Lake Edko and Abu Hummus fish farm revealed the presence of only five compounds, heptachlorepoxide, p,p- DDE, dieldrin, p,p-DDD and endrin ketone, with concentrations exceed the permissible limits proposed by WHO [26-28] and EMOHR [19]. Fortunately, Egyptian people did not use the water of both Edko lake and fish farm in drinking.
References
- EPA. Environmental Protection Agency Aquatic biodiversity. 2009.
- Schulte-Oehlmann U, Oehlmann J, Keil F. Before the Curtain Falls: Endocrine-Active Pesticides - A German Contamination Legacy. Whitacre DM, editors. In: Reviews of Environmental Contamination and Toxicology. 2011; 213: 137-159.
- Badawy MI. Use and impact of pesticides in Egypt. International Journal of Environmental Health Research. 1998; 8: 223-229.
- Shakoori AR, Mughal AL, Iqbal MJ. Effect of sublethal doses of fenvalerate (a synthetic pyrethroid) administered continuously for four weeks on the blood, liver and muscles of a freshwater fish Ctenopharyngodon idella. Bull Environ Contam Toxicol. 1996; 57: 487-494.
- Moreira SM, Guilhermino L. The use of Mytilus galloprovincialis acethylcholinesterase and gluthatione-S-transferase activities as biomarkers of environmental contamination along the northwest Portuguese coast. Environmental Monitoring and Assessment. 2005; 105; 309-325.
- Singh KP, Malik A, Mohan D, Takroo R. Distribution of persistent organochlorine pesticide residue in Gomti river, India. Bulletin of Environmental Contamination and Toxicology. 2005; 74: 146-154.
- Aulakh JS, Malik AK, Kaur V, Schmitt-Kopplin P. A review on Solid Phase Micro Extraction-High Performance Liquid Chromatography (SPME-HPLC) analysis of pesticides. Crit. Rev. Anal. Chem. 2005; 35: 71-85.
- Lehotay SJ, Mastovska´ K, Yun SJ. Evaluation of two fast and easy methods for pesticide residue analysis in fatty food matrixes. J AOAC Int. 2005; 88: 630-638.
- Hill ARC, Reynolds SL. Guideline for in-house validation of analytical methods for pesticide residues in food and animal feeds. Analyst. 1999; 124: 953-958.
- Tomkins BA, Merriweather R, Jenkins RA. Determination pf eight organochlorine pesticides at low nanogram/l concentrations in ground water using filter disk extraction and gas chromatography J. Assoc of Anal. 1992; 75: 1091-1099.
- PAM. Pesticide Analytical Manual, US. Food and Drug Administration. US. Department of Health and Human Service. Washington, DC, USA. 1994.
- EPA. Environmental Protection Agency. Method 525.1, revision 2.2. Method for the determination of organic compounds in drinking water. Fedral Register. 1994; 56: 30272.
- IEH. Web Report W20. Leicester, UK, MRC Institute for Environment and Health. 2005.
- USP. United States Pharmacopeia 33 (2010) 256. United States Pharmacopeia convention. Inc., Rockville. MD20852 USA. 2010.
- Matsumura MM, Margulius SP, Saligman AM. Environmental Toxicology of pesticides. Academic Press New York. 1972; 30.
- Bowma BT, Sans WW. Stability of parathion and DDT in dilute iron solution. J Environ Sci Health. 1980; 15: 233.
- Laymann WJ, Reehl WF, Rosenblatt DH. Handbook of chemical property Estimation Methods. Amer Chem Soc USA. Washington DC. 1990.
- Velocgaleti R, Burns PK, Gill M, Prothro J. Impact of current good manufacturing practices and emission regulations and guidance on the discharge of pharmaceutical chemicals into the environment from manufacturing use and disposal. Environ. Health Perspect. 2002; 110: 213- 220.
- EMOHR. Egyptian Ministry of Health Regulation report NO 92. In Arabic. 2013.
- EEC. European Economic Community, Drinking water directive. Official Journal N 229/11 Directive 80/778. 1988.
- WHO. World Health Organization, Guidelines for Drinking Water Quality 2nd Edition. Vol-1, Geneva. Switzerland. 1993.
- Sandín-España P, Sevilla-Morán B. Pesticide Degradation in Water. Rathore HS, Nollet LML, editors. In: Pesticides Evaluation of Environmental Pollution. CRC Press Taylor & Francis Group 6000 Broken Sound Parkway NW, Suite 300 Boca Raton. FL 33487-2742. 2012; 4: 79-119.
- Beltran J, Lopez F, Hernandez F. Solid-phase micro-extraction in pesticide residue analysis. J. Cromatogr. 2000; 885: 389-404.
- Carabias-Martínez R, Rodríguez-Gonzalo E, Herrero-Hernández E, Hernández-Méndez J. Simultaneous determination of phenyl- and sulfonylurea herbicides in water by solid- phase extraction and liquid chromatography with UV diode array or mass spectrometric detection. Anal. Chim. Acta. 2004; 517: 71-79.
- Sabik H, Jeannot R, Rondeau B. Multiresidue methods using solid-phase extraction techniques for monitoring priority pesticides, including triazines and degradation products, in ground and surface waters. J Chromatogr. 2000; 885: 217-236.
- WHO. World Health Organization expert panel report, cited in Edwards, Rob. “Industry denies dangers of particle pollution”. New Scientist. 1995; 4.
- Colborn T, Dumanoski D, Myers JP. Our Stolen Future, 306 pp. New York: Penguin Books USA Inc. 1996.
- Sanches SM, Silva CHTP, Campos SX, Vieira EM. Pesticidas e seus respectivos riscos associados a` contaminacoda ´gua (Pesticides and respective risks associated to water contamination). Pesticidas Ecotoxicole Meio Ambiente. 2003; 13: 53-58.