
Abstract
A thin patient with bradykinesia and gait disturbance being suffered from recurrent dyspnea and a mood disorder would be promptly considered of the diagnosis of Perry syndrome. This disorder is known as TDP-43 proteinopathy caused by DCTN1 gene mutation is clinically characterized by autosomal dominant inheritance. Also, we could find that the [18F] FDG PET showed cortical atrophy and glucose hypometabolism in the subgenual prefrontal cortex, anterior cingulate cortex and insula from this patient. The marked loss of [18F] FP-CIT binding in the dorsal and ventral striata would be checked, too.
Keywords: Perry syndrome; Central hypoventilation; TDP-43 proteinopathy; DCTN1 gene; [18F] FDG PET, [18F] FP-CIT; Gene study
Introduction
Perry syndrome is a progressive brain disease with four major characteristics. Those are composed of parkinsonism, weight loss, hypoventilation as well as psychiatric changes [1,2]. Perry syndrome is a rare hereditary disease, characterized by parkinsonism, depression, central hypoventilation and weight loss. This disorder is known as TDP-43 proteinopathy with mutations in the dynactin gene (DCTN1) [3]. Depression and parkinsonism tend to occur early whereas hypoventilation and severe weight loss manifest later.4,5 In particular, depression is often accompanied by other psychiatric features, such as apathy, character changes and social withdrawal suggesting involvement of frontal cortex [4,5].
Case
Clinical History, Neurological Examination and Laboratory Finding
A 54-year-old woman who presented with bradykinesia and shuffling 2 years ago. She had developed depression for several years. And her sister and mother were diagnosed with Parkinson’s disease with the ventilator equipment. Finally, one of them died from respiratory failure (Figure 1). This patient showed gait problem, bradykinesia, rigidity but not tremor. As her disease worsened, the physical examination revealed an average built female except her weight checking just 38Kg. However, her memory, calculation or insight was so good. Moreover, her cerebellar function and postural balance as well as eye ball movement did not reveal any abnormal finding. After 2 years, she rushed into the emergency room for respiratory failure. We checked her arterial blood gas analysis showing marked hypercarbia (pCO2=74 mmHg). Respiratory monitoring showed frequent spells of apnea, each lasting over 20 sec. Nerve conduction and EMG studies were normal. Also, she had showed anxiety and social withdrawal symptom with depression from her disease. There was not definite tremor of all four extremities and with normal tendon jerks. Both plantar muscles were flexor. Fundus examination was shown unremarkable. Other nervous system and systemic examination was unremarkable. Routine laboratory evaluation was normal including routine complete blood count and chemistry panel. Also, he did not show abnormal thyroid function, Vitamin B12 level, folate level, methylmalonic acid level, homocysteine level or tumor marker level for CA125, AFP, CEA and CA 19-9. The patient was negative for autoimmune disease work-up, including anti-dsDNA antibody, anti-cardiolipin antibody, anti-sm antibody, anti-SS-A/B, HLA B27 antibody or HLA B51 for Bechet’s Disease, hepatitis B and C, as well as HIV. The chest X-ray showed non-specific features. Furthermore, Serum ceruloplasmin level, 24-hour urinary copper excretion, serum copper or and Peripheral blood film did not reveal abnormal finding.
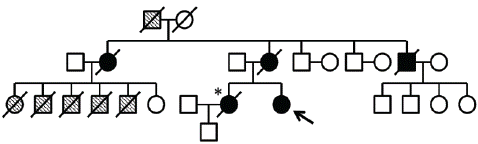
Figure 1: The arrow indicates this patient. The sister of the proband is indicated by *: she suffered from young-onset parkinsonism and died of respiratory failure after years of ventilator support. Black circles and squares indicate family members who manifested parkinsonism. Hatched circles and squares indicate family members who died of unidentified causes at young age.
Neuroradiologic Finding, Neuropsychology Test Finding and Gene Test
She was evaluated with [18F] FP-CIT PET (18F-fluorinated N-3-fluoropropyl-2-ß-carboxymethoxy-3-β-(4-iodophenyl) nortropane), [18F] FDG (fluorodeoxyglucose) PET and brain 1.5T MR as well as neuropsychological test. MR showed significant cortical thinning in the right subgenual prefrontal cortex, anterior cingulate cortex, insula, medial part of superior frontal gyrus, right and left middle cingulate cortex, and supplementary motor area (p<0.01, Figure 2).
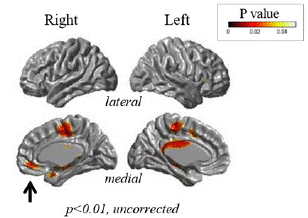
Figure 2: Cortical thickness measurements showed significant atrophy in the subgenual prefrontal cortex (arrow, p<0.01,
uncorrected), consistent with [18F] FDG PET findings.
[18F] FP-CIT PET showed marked loss of dopamine transporters. (Figure 3) In addition, [18F] FDG-PET showed significant glucose hypometabolism in the anterior cingulate cortex, orbitofrontal cortex, pars opercularis of the inferior frontal gyrus, and inferior parietal lobule. (p<0.05, FDR corrected, 100-voxel level, one sample t-test). (Figure 4).
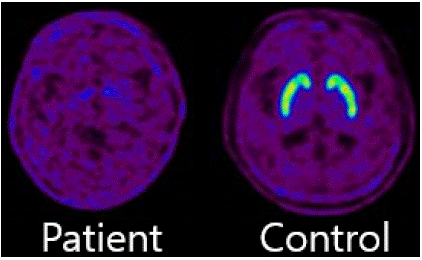
Figure 3: [18F] FP-CIT PET image of the patient (left) showed almost complete loss of ligand binding to the dopamine transporter across all regions of the striatum, compared to the image of an age-matched healthy subject (right). These [18F] FP-CIT PET findings are in sharp contrast to the relatively mild parkinsonism (off UPDRS III score = 36), manifested in this patient. This apparent discrepancy may be due to the combined loss of dopaminergic and serotonergic terminals in the striatum, as FP-CIT exhibits also affinity for the serotonin transporter (SERT).
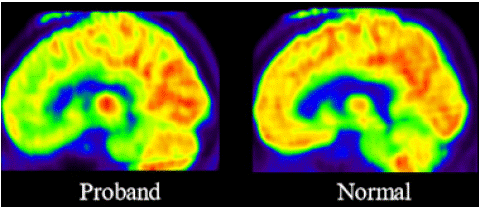
Figure 4: [18F] FDG PET image of the patient (left) was compared with that of age-matched healthy subject (right). Quantitative analysis of [18F] FDG PET showed significant glucose hypometabolism in the subgenual prefrontal cortex, anterior cingulate cortex, insular, caudate nucleus and putamen (p<0.01).
Her Mini-Mental State Exam (MMSE) score was 28 out of 30 and orientation with attention was normal but working memory was normal. Furthermore, word fluency and inhibitory control ability were abnormal. Also, confrontational naming and calculation ability was shown prompt and normal and immediate recall, delayed recall including recognition was good. Korean-Beck Depression score was 23 out of 63 to demonstrate significant depression.
We identified gene test and identified the mutation (Gly67Asp) at c.518G>A in DCTN1, the dynactin gene, resulting in loss of its functional properties by changing one amino acid to other. Gene testing identified a novel mutation at c.518G>A (p.Gly67Asp) in DCTN1, consistent with Perry syndrome (Figure 5)
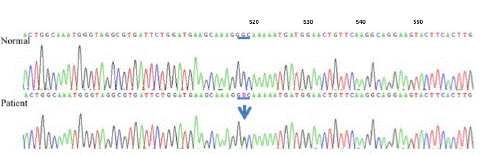
Figure 5: Electropherogram demarked the mutation in DCTN1. Arrows would be the locus of heterozygous point mutations.
Discussion
It was identified as the mutation of TDP-43 proteinopathy in the dynactin gene [1,3]. Though clinical symptoms, we could check typical features of Perry syndrome like Parkinsonism, central hypoventilation, depression and weight loss. Especially, depression and parkinsonian feature may occur early whereas hypoventilation and severe weight loss tend to develop later.1 Furthermore, depression is often accompanied by other psychiatric features, such as apathy, character changes and social withdrawal, suggesting involvement of frontal cortex [1,6]. And [18F] FDG-PET CAT scan of this patient showed glucose hypometabolism for insula
including ant. paralimbic area, subgenual prefrontal cortex, orbitofrontal cortex as wellas cingulate cortex [6-8]. (Figure 6) In particular, olfactocentric paralimbic cortex revealed thinned gray matter volume [6-8]. Thus, these findings may be attributed to loss of serotonergic neurons as well as dopaminergic neurons neuroimaging studies in primary and secondary depression have shown glucose hypometabolism in the subgenual and orbitofrontalcortex [6-8].
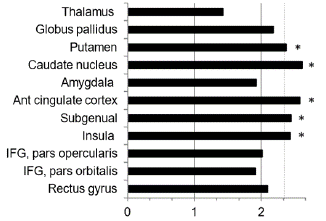
Figure 6: Bar graph of z-scores for the FDG uptake would explain the difference of glucose metabolism. Y-axis represent cortical and subcortical regions. X-axis is that higher z-score means lower glucose metabolism. The broken line indicates p=0.01, which corresponds to z-score of 2.33. IFG: inferior frontal gyrus. *Regions with significant glucose hypometabolism
Moreover, some of the marked loss of [18F] FP-CIT binding in the dorsal and ventralstriata may be caused by loss of seroFigure tonergic neurons and dopaminergic neurons since[18F] FP-CIT has a relatively high affinity to serotonin [6-9].
However, previous pathological studies have reported relatively sparing of the cerebral cortex in this syndrome [5].
Finally, we could suggest that the depressive symptom with Perry syndrome might beassociated with decreased neural activity in the subgenual prefrontal cortex and related paralimbic regions, possibly accompanied by severe serotonergic denervation.
Author Statements
Acknowledgments
We would like to express our gratitude to the patient and her family for the participation in this case study.
Authorship Contributions
Medical Practices: E.J.C., Concept: E.J.C.; Design: E.J.C., Data Collection or Processing: E.J.C., D.G.L., DS.S.; Analysis or Interpretation: E.J.C., DS.S.; Literature Search: DS.S., D.G.L., E.J.C., Writing: D.G.L., E.J.C.
Funding
The authors declared that this study received no financial support.
Availability of Data and Materials
The datasets supporting the conclusion of this article is included within the article and its figure file.
Consent for Publication
Written informed consent was obtained from the patient before her death and from her immediate family after her death for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-Chief of this journal.
Competing Interests
No author states to have any competing interest.
References
- Tacik P, Fiesel FC, Fujioka S, Ross OA, Pretelt F, Castañeda Cardona C, et al. Three families with Perry syndrome from distinct parts of the world. Parkinsonism Relat Disord. 2014; 20: 884-8
- Aji BM, Medley G, O’Driscoll K, Larner AJ, Alusi SH. Perry syndrome: a disorder to consider in the differential diagnosis of Parkinsonism. J Neurol Sci. 2013; 330: 117-8.
- Mishima T, Koga S, Lin WL, Kasanuki K, Castanedes-Casey M, Wszolek ZK, et al. Perry Syndrome: A Distinctive Type of TDP-43 Proteinopathy. J Neuropathol Exp Neurol. 2017; 76: 676-682.
- Jaroslaw Dulski, Takuya Konno, Zbigniew Wszolek. DCTN1-Related Neurodegeneration. Gene Reviews. 2010.
- Chung EJ, Hwang JH, Lee MJ, Hong JH, Ji KH, Yoo WK, et al. Expansion of the clinicopathological and mutational spectrum of Perry syndrome. Parkinsonism Relat Disord. 2014; 20: 388-93.
- Oh M, Kim JS, Kim JY, Shin KH, Park SH, Kim HO, et al. Subregional patterns of preferential striatal dopamine transporter loss differ in Parkinson disease, progressive supranuclear palsy, and multiple-system atrophy. J Nucl Med. 2012; 53: 399-406.
- Ziebell M, Holm-Hansen S, Thomsen G, Wagner A, Jensen P, Pinborg LH, et al. Serotonin transporters in dopamine transporter imaging: a head-to-head comparison of dopamine transporter SPECT radioligands 123I-FP-CIT and 123I-PE2I. J Nucl Med. 2010; 51: 1885-1891.
- Borgers AJ, Alkemade A, Van de Giessen EM, Drent ML, Booji J, Bisschop PH, et al. Imaging of serotonin transporters with [123I] FP-CIT SPECT in the human hypothalamus. EJNMMI Res. 2013; 3: 34.
- Lee CS, You CG, Kim KA, Kim JS. Aging effects of [18F] FP-CIT PET measures in the striatum of drug-naive, de novo PD patients: Comparison with normal subjects. Neurology. 2010; 74: A249.