
Abstract
CJD is a fatal neurodegenerative disease with an estimated prevalence of one case per million. It often occurs sporadically but familial and iatrogenic cases occur. CJD is caused by the accumulation of pathologic prion proteins (PrPSc) in neuronal tissue leading to neuronal loss. It, typically, presents with rapidly progressive dementia, dysarthria, cerebellar ataxia, and myoclonus, culminating in a kinetic mutism and death. There are at least five different phenotypes for its initial clinical presentation. This paper will describe a case of CJD, explore the clinical presentations of CJD, and discuss a systematic approach to diagnosing CJD.
Keywords: Creutzfeldt - Jakob disease; Prion disease; Sub-acute ataxia; Myoclonus; Dementia
Introduction
Creutzfeldt-Jakob Disease (CJD) a rare and fatal neurodegenerative caused by accumulation of a pathological prion protein (PrPSc) in neuronal tissue leading to neuronal loss, astrocytic gliosis and spongiform changes [1]. Most cases of CJD are sporadic but familial and iatrogenic cases (e.g., transmission through human pituitary growth hormone, dura mater grafts and corneal grafts) do occur [1]. CJD classically presents with rapidly progressive dementia, dysarthria, cerebellar ataxia, myoclonus culminating in akinetic mutism and death [2]. However, there is phenotypic heterogeneity in its presentation [3]. This paper explores clinical presentations of CJD and discusses investigational techniques used to diagnose CJD in the hospital setting.
Case Presentation
A 69 year-old man with history of diabetes mellitus, coronary artery disease status post percutaneous coronary intervention complicated by cardiac arrest and subsequent pacemaker placement, obstructive sleep apnea, hypertension, and atrial fibrillation presented to the emergency department (ED) at the Veterans Affairs (VA) hospital in San Diego, California with lower extremity edema. He was prescribed furosemide and discharged home.
Two days later, he returned to the ED with slurred speech and gait imbalance. He had normal vital signs, mild dysarthria, decreased attention, left-sided pronator drift, tremor, abnormal coordination, peripheral neuropathy and unsteady, broad-based gait with decreased left arm swing. CT head was unrevealing. CT angiogram showed bilateral moderate-to-severe proximal internal carotid artery stenosis. Transthoracic echocardiogram revealed declined ejection fraction from 61% one year prior to 39%. Brain MRI was not performed due to patient’s pacemaker. His symptoms improved with intravenous hydration. He was discharged home.
Two weeks later, he returned to the hospital with left-sided weakness, confusion and word-finding difficulties. Examination revealed impaired language output, child-like affect and truncal ataxia. Reversible causes of dementia were ruled out (Table 1). The neurology consultants recommended genetic testing to rule out heritable ataxias. He was discharged to a skilled nursing facility with scheduled outpatient neurology follow-up.
![]()
Metabolic Panel
Values
Units
- Sodium
139
mMol/L
- Potassium
4.0
mMol/L
- Chloride
101
mMol/L
- CO2
25
mMol/L
- Anion Gap
13
mMol/L
- Blood urea nitrogen
17
mg/dL
- Creatinine
0.86
mg/dL
- Glucose
92
mg/dL
- Calcium
9.4
mg/dL
- PO4
3.7
mg/dL
- Magnesium
2.2
mg/dL
- Aspartate aminotransferase
26
IU/L
- Alanine aminotransferase
28
IU/L
- Alkaline phosphatase
62
IU/L
- Total bilirubin
1.7
mg/dL
- Albumin
4.5
g/dL
Lipid Panel
- Cholesterol
155
mg/dL
- Calculated LDL
91
mg/dL
- HDL
38
mg/dL
- Triglyceride
129
mg/dL
Urine toxicology
Negative
Miscellaneous
- TSH
2.7
- Copper
113
mcg/dL
- Ceruloplasmin
30
Mg/dL
- Homocysteine
10.61
umol/L
- Methylmalonic acid
0.16
umol/L
- B12
1457
pg/ML
- Alpha-Tocopherol (Vit E)
16.0
mg/L
Microbiology
- RPR
Non-reactive
- HIV
Negative
- Lyme EIA screen
Negative
- Rabies Ab
0.00
EU/mL
- Clostridium difficle Tox B gene
Positive
Cerebral Spinal Fluid
- HSV 1 DNA
Not detected
- HSV 2 DNA
Not detected
- CMV DNA
Not detected
- VDRL
Non-reactive
- Cocci CF
Negative
- Mycology
Negative
- Tau
3382
pg/mL
- 14-3-3
Positive
Table 1: Laboratory data during hospitalization.
Three weeks later, he presented to the ED with declining speech production, startle myoclonus and somnolence requiring endotracheal intubation for airway protection. Cerebrospinal fluid (CSF) testing for infectious etiologies and pan-CT imaging were unrevealing. EEG showed generalized, occasionally rhythmic, medium to high amplitude theta and delta waves without evidence of seizures or epileptiform discharges.
The patient's hospitalization was complicated by respiratory failure, myocardial infarction, atrial flutter with rapid ventricular response, sepsis, Clostridium difficile colitis and progressive neurologic decline. In accordance with his family’s wishes, he was transitioned to comfort care and passed away approximately two months after his initial presentation.
Given the sub-acute decline in his neurologic status with startle myoclonus and otherwise unrevealing work-up, a diagnosis of CJD was suspected. CSF analysis revealed 14-3-3 and tau proteins (3382pg/mL). Final postmortem neuropathology of the left and right frontal cortices and white matter showed numerous variably-sized, occasionally coalescent, vacuoles within the neuropil and spongiform change accompanied by occasional loss of neurons and gliosis, consistent with CJD. The tissue was submitted to the National Prion Disease Pathology Surveillance Center where immunostaining with 3F4, monoclonal antibody to the prion protein, confirmed a diagnosis of prion disease, likely CJD (Figure 1).
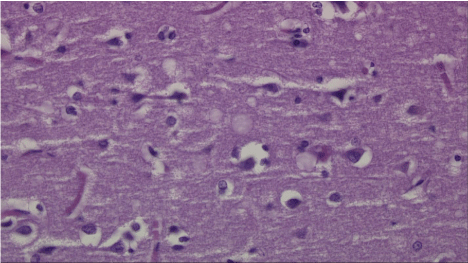
Figure 1: Neuropathological Microscopic Description.
Sections of the right and left frontal cortex and white matter show numerous,
variably sized and occasionally coalescent vacuoles within the neuropil. This
is consistent with Creutzfeldt-Jakob Disease (CJD). The spongiform change
is accompanied by occasional loss of neurons and gliosis. After histologic
review of H&E stained sections, the material was submitted for additional
testing to The National Prion Disease Pathology Surveillance Center
(NPDPSC) NPDPSC (https://www.cjdsurveillance.com). Their consulting
diagnosis read as follows: “Immunostaining with 3F4, the monoclonal antibody
to the prion protein, reveals granular deposits as seen in prion disease. This
finding establishes the diagnosis of prion disease, likely Creutzfeldt-Jakob
disease.” A final neuropathological diagnosis of CJD was eventually made.
Figure and pathological interpretation courtesy of Dr. Peter Kobalka and Dr.
Subhojit Roy at the VA Hospital in San Diego, California.
Discussion
CJD classically presents with rapidly progressive dementia, cerebellar ataxia, speech difficulties and myoclonus [2], but there is clinical heterogeneity in its initial presentation. A subset of patients may present with an isolated cerebellar syndrome with cognitive decline seen weeks to months later [3]. Some may present with an isolated visual disturbance which progresses to cortical blindness. Others may present with psychiatric symptoms prior to developing dementia and neurologic decline [1,3] (Table 2).
![]()
Subtype
Presentation
Classic CJD
Initially presents with cognitive symptoms (e.g., amnesia, language impairment), executive dysfunction and ataxia. Does not present with visual disturbance and has a survival time of less than 3 months.
Heidenhain
Initially presents with diplopia, blurred vision, cortical blindness and/or visual hallucinations and has a survival time of less than 4 months.
Oppenheimer-Brownell
Initially presents with ataxia in the absence of other symptoms. Median age of onset is 67 years-old. There is an absence of periodic sharp wave complexes (PSWCs) on EEG.
Cognitive
Presents with dementia, memory and language impairment, disorientation, executive dysfunction and/or disorientation. Does not present with ataxia or visual disturbance at onset. Survival time is greater than 4 months.
Affective
Initially presents with depression, mood liability and/or anxiety with ataxia with an age of onset less than or equal to 65 year-old. Survival time is greater than 6 months.
1Adapted from: “Characteristics of Established and Proposed Creutzfeldt-Jakob Disease Variants,” by Abbleby et al., 2009. In the journal JAMA Neurology.
Table 2: Appleby and colleagues (2009) performed a retrospective analysis of 88 patients with definite and probable CJD and characterized the following five CJD subtypes1.
Given this heterogeneity, it is important to consider CJD when evaluating patients with rapidly progressive neurologic signs and symptoms. Basic labs should be obtained to rule out reversible causes. If reversible causes cannot be identified, then other causes of rapidly progressive dementia should be considered including paraneoplastic neurologic syndromes, auto-immune diseases, central nervous system vasculitis and infections such as HIV, neurosyphilis, herpes simplex virus encephalitis, Lyme disease, rabies and Whipple's disease. Once these have been ruled out, one must consider CJD [4].
In our case, the patient initially presented with sub-acute ataxia and then developed dementia. The differential diagnosis for subacute ataxia is broad and includes atypical infections, auto-immune disorders, primary or metastatic tumors, paraneoplastic cerebellar degeneration, alcohol abuse, vitamin deficiencies and other systemic disorders (Table 3).
![]()
Disease
Diagnostic
Atypical infectious agents
Progressive multifocal leukoencephalopathy
PCR detection of JC virus DNA
Prion disease (eg CJD)
Brain biopsy, CSF 14-3-3
Whipple disease
Jejunal biopsy Tropheryma whippelii
Autoimmune disease
Multiple sclerosis
Brain MRI, CSF oligoclonal bands
Acute disseminated encephalomyelitis
Brain MRI
Miller fisher syndrome
Anti GQ1b antibodies
Glutamic acid decarboxylase antibody associated ataxia
Anti-GAD antibodies
Celiac disease
TTGA IgA +/- IgA level
Hashimotos encephalopathy
Anti-thyroid antibody
Sarcoidosis
Biopsy of site, contrast MRI, CSF analysis
Primary or metastatic tumor
Neuroimaging
Paraneoplastic cerebellar degeneration
Neuroimaging, CSF analysis
Wernicke encephalopathy
Thiamine deficiency, exam with deficits in mentation, oculomotor function and ataxia
Vitamin E deficiency
Vitamin B12 deficiency
Copper deficiency
Systemic disorders
Acquired hepatocerebral degeneration
Brain MRI
Hypothyroidism
TSH
Hypoparathyroidism
PTH
Table1 Adapted from “Overview of cerebellar ataxia in adults” by Peter Todd, in UpToDate, December 14, 2015.
Table 3: Causes of sub-acute ataxia1.
A definitive diagnosis of CJD requires brain biopsy with neuropathology demonstrating neuronal loss, astrocytic gliosis, spongiform changes and presence of PrPSc [1]. If brain tissue cannot be obtained, the World Health Organization (WHO) criteria can guide the diagnosis of possible versus probable CJD [5] (Table 4).
![]()
Sporadic CJD
1. Diagnosed by standard neuropathological techniques
2. And/Or immunocytochemically
3. And/Or Western blot confirmed protease-resistant prior protein (PrP)
4. And/Or presence of scrapie-associated fibrils
Probable Sporadic CJD
1. Progressive dementia
2. At least 2 of the following 4 symptoms:
a. Myoclonus
b. Pyramidal/extrapyramidal
c. Visual or cerebellar
d. Akinetic mutism
3. Positive EEG (periodic epileptiform discharges)
4. And/Or positive 14-3-3 protein result and < 2 year disease duration
5. Routine investigations do not suggest an alternative diagnosis
Possible Sporadic CJD
1. Progressive dementia
2. At least 2 of the following 4 symptoms:
a. Myoclonus
b. Pyramidal/extrapyramidal
c. Visual or cerebellar
d. Akinetic mutism
3. No supportive EEG
1Global Surveillance, Diagnosis and Therapy of Human Transmissible Spongiform Encephalopathies: Report of a WHO Consultation. Geneva, Switzerland, 9-11 February 1998.
Table 4: World Health Organization Criteria for Diagnosis of CJD (1998)1.
EEG in patients with CJD often shows deterioration in the normal background rhythms and periodic sharp wave complexes (PSWCs) with a specificity ranging from 66% to 91% [6]. The absence of PSWCs does not exclude a diagnosis of CJD; therefore, EEG testing should be repeated regularly [1].
Tau and 14-3-3 proteins are released into the CSF following neuronal damage. The detection of CSF 14-3-3 protein is 97% sensitive and 87% specific for CJD [7]. Since these proteins are released when neurons are damaged, false positives due to other diseases including meningoencephalitis, multi-infarct dementia, hypoxic brain injury, Alzheimer’s disease, Lewy body dementia, and intracerebral malignancy may occur [8]. Given this, CSF 14-3-3 is only a useful test when there is a high pre-test probability for CJD and other etiologies have been ruled out. Along with the elevated CSF 14-3-3 proteins, tau proteins may also be detected in sCJD, but, these proteins are also general markers for neuronal injury and may be falsely elevated [9].
Recently DWI and FLAIR MRI brain sequences are being used for diagnosing CJD. The technique can differentiate between CJD and non-prion causes of rapidly progressive dementia (npRPD) with sensitivity and specificity for CJD of 96% and 93%, respectively. In CJD, gray matter hyper-intensities are observed in neocortical, limbic and/or subcortical areas, whereas neocortical hyper-intensity is not observed in npRPD [10].
Conclusion
Recognizing nuances in presentation and testing is important when considering a diagnosis of CJD as there is no intervention to slow the progression. The goal of management should emphasize family counseling and education, comfort care and discourage the use of invasive measures that unnecessarily prolong life. Family members can be informed of CJD research protocols at referral centers such as Case Western Reserve University and University of California San Francisco, which can enroll patients by receiving clinical and neuroimaging records as well as blood or CSF samples when available.
Acknowledgements
We thank Dr. Sara Patrawala, Dr. Peter Kobalka and Dr. Subhojit Roy their assistance with this case.
References
- Knight RSG & Will RG. Prion Diseases. Journal of Neurology, Neurosurgery, and Psychiatry. 2004; 75: i36-i42.
- Imran M & Mahmood S. An overview of human prion diseases. Virology Journal. 2011; 8: 559-568.
- Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT & Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. JAMA Neurology. 2009; 66: 208-215.
- Lee R, Buckley C, Irani SR, Vincent A. Autoantibody testing in encephalitis. Practical Neurology. 2012; 12: 4-13.
- Global Surveillance, Diagnosis and Therapy of Human Transmissible Spongiform Encephalopathies: Report of a WHO Consultation, Geneva, Switzerland, 9-11 February 1998.
- Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S. & Kretszchmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Annals of Neurology. 2004; 56: 702-708.
- Lemastra AW, van Meegen MT, Veryling JP, Meigerink PH, Jansen GH, Baas F, et al. 14-3-3 testing in diagnosing Creutzfeldt-Jakob disease: a prospective study in 112 patients. Neurology. 2000; 55: 514-516.
- Green AJE. Use of 14-3-3 in the diagnosis of Creutzfeldt-Jakob disease. Biochemical Society Transactions. 2002; 30: 382-386.
- Rosenblum MH, Atria A. The evaluation of rapidly progressive dementia. Neurologist. 2011; 17: 67-74.
- Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76: 1711-1719.