
Abstract
Patients with Diabetes Mellitus (DM) have accelerated atherosclerosis, which is the main essential factor contributing to the high risk of atherothrombotic events in these patients. Atherothrombotic complications are the principal cause of morbidity and mortality in patients with DM. Both atherosclerosis and the increased risk of thrombotic vascular events may result from dyslipidaemia, endothelial dysfunction, platelet hyperreactivity, an impaired fibrinolytic balance, and abnormal blood flow. Platelets of DM patients are characterised by dysregulation of several signalling pathways causing increased adhesion, activation and aggregation. Platelet function of patients with DM is complicated by several mechanisms, such as hyperglycaemia, insulin deficiency and resistance, associated metabolic conditions, and cellular abnormalities. The present manuscript purposes to provide a review on the up-to-date status of data on platelet abnormalities that characterise patients with DM.
Introduction
The most important factor that contributes to the increased risk of atherothrombotic events in patients with Diabetes Mellitus (DM) is accelerated atherosclerosis. Cardiovascular disease, mainly Coronary Artery Disease (CAD), including Acute Coronary Syndrome (ACS), is the first cause of morbidity and mortality in these patients [1]. It is reported 20 years ago that DM patients without any history of CAD have the same cardiac mortality risk as non-DM patients with a history of Myocardial Infarction (MI) [2]. Furthermore, cardiovascular disease has equally poor prognosis in patients with DM as they have a higher risk of complications and recurrent atherothrombotic events than non-DM patients [3]. In fact, in an ACS scenario, DM is a strong independent predictor ischemic events recurrence, including mortality [4]. After all, the presence of comorbidities that have negative impact on ACS outcomes is higher in DM patients [5]. Several factors are involved to the prothrombotic condition of patients with DM, such as the following: increased coagulation, impaired fibrinolysis, endothelial dysfunction and platelet hyper reactivity [6,7]. The latter is of specific importance, since platelets play a pivotal role in the formation, development and sustainment of thrombi [8]. Platelets of patients with DM are characterised by dysregulation of several signalling pathways, and they are hyper reactive with intensified adhesion, activation and aggregation [6,9-12]. Such a hyperreactive platelet may result to the higher part of DM patients with insufficient response to anti platelet agents compared with non-DM subjects [13,14]. Several metabolic and cellular abnormalities provoke multiple mechanisms that play a role in the increased platelet reactivity observed in patients with DM. These mechanisms can be joined into the following aetiopathogenic categories [15]: a) hyperglycaemia, b) insulin deficiency and resistance, c) associated metabolic conditions, and d) other cellular abnormalities (Table 1).
![]()
MECHANISMS CONTRIBUTING TO PLATELET DYSFUNCTION IN PATIENTS WITH DM
Hyperglycaemia
Deficient Insulin Action
Associated Metabolic Conditions
Other Cellular Abnormalities
Platelet
Endothelial Dysfunction
Increased
Impaired response to NO and PGI2
Obesity
Increased platelets turnover
Increased production of TF
P-select in expression
Osmotic effect
IRS-dependent factors:
Dyslipidaemia
Increased intracellular Ca++
Decreased NO and PGI2 production
- increased intracellular Ca++
- degranulation
Activation of PKC
Inflammation
Upregulation of P2Y12 signalling
Decreased membrane fluidity by glycation of surface protein
Oxidative stress
Increased
P-selectin and GP expression
Table 1: Mechanisms contributing to platelet dysfunction in patients with diabetes mellitus (DM). Ca++: calcium, GP: glycoprotein, IRS: insulin receptor substrate, NO: nitric oxide, PGI2: prostacyclin, PKC: protein kinase C, TF: tissue factor.
The aim of this paper is to provide an up-to-date review on the characteristic profile of the “diabetic” platelet.
Hyperglycaemia
Diabetes is a metabolic disease characterised by hyperglycaemia caused by defects in insulin secretion, insulin action, or both [16]. Hyperglycaemia not only describes DM but also plays an independent and significant role in the blood irregularities leading to a prothrombotic state in DM patients [17,18].
Platelets from patients with type-2 DM have greater expression of adhesion molecules. A study by Eibl et al proved that platelets from DM patients have an increased expression of platelet activation markers compared with an age-matched non-diabetic control group [19]. On the same wavelength, recently Soma et al used electron microscopy and flow cytometry to show the same thing. The exact association between hyperglycaemia and platelet activation markers (CD31, CD49b, CD62P and CD63) was found when there was a significant decrease in their expression after improving metabolic control during 3 months. Furthermore, an important correlation between CD62P, CD63 and HbA1C levels reported. These data recommend that amelioration of glycaemic control may have a positive effect on platelet adhesion and following activation [19]. Platelet surface receptors are also highly expressed in DM patients. Especially, expression of glycoprotein (GP) Ib and GP IIb/IIIa, which respectively facilitate the binding with the von Willebrand factor leading to platelet-fibrin interaction, is increased in DM patients [11,12,14].
Acute hyperglycaemia consequences in amplified platelet activation, as known by high levels of surface adhesion molecules such as P-selectin [20] and soluble markers of platelet activation (e.g. soluble P-selectin and CD40 ligand) [21,22]. In addition, levels of HbA1C and fasting glucose have been linked to P-select in expression in type-2 DM patients undergoing coronary angioplasty [23], which recommends again that an improved glycaemic profile may reduce platelet reactivity. Some clinical data, along with the above stated laboratory findings, support the idea that anti-diabetic therapy improves the metabolic control and benefits DM patients with atherothrombotic complications. The intensive glucose-lowering treatment compared to standard treatment in a randomised trial showed that mortality in the intensive treatment group treated with insulin was significantly lower than in the control group treated with standard therapy [24]. Moreover there was no difference in mortality or morbidity among three different glucose-lowering strategies [25]. It was also proved that the profit of reducing glucose levels is independent of the way it is achieved since the glucose-lowering levels were similar among the study groups of the previous study. However, the optimal blood glucose level rests unknown. On the other hand, an extreme lowering of glucose has been confirmed to be unsafe in several trials, which randomised DM patients to receive an intensive glucose-lowering regimen or a standard regimen. The trial was stopped prematurely after 3.5 years of follow-up due to an increased mortality in the intensive therapy group [26,27].
Several mechanisms have been suggested to contribute to the increased platelet reactivity caused by hyperglycaemia, including the following (Figure 1).
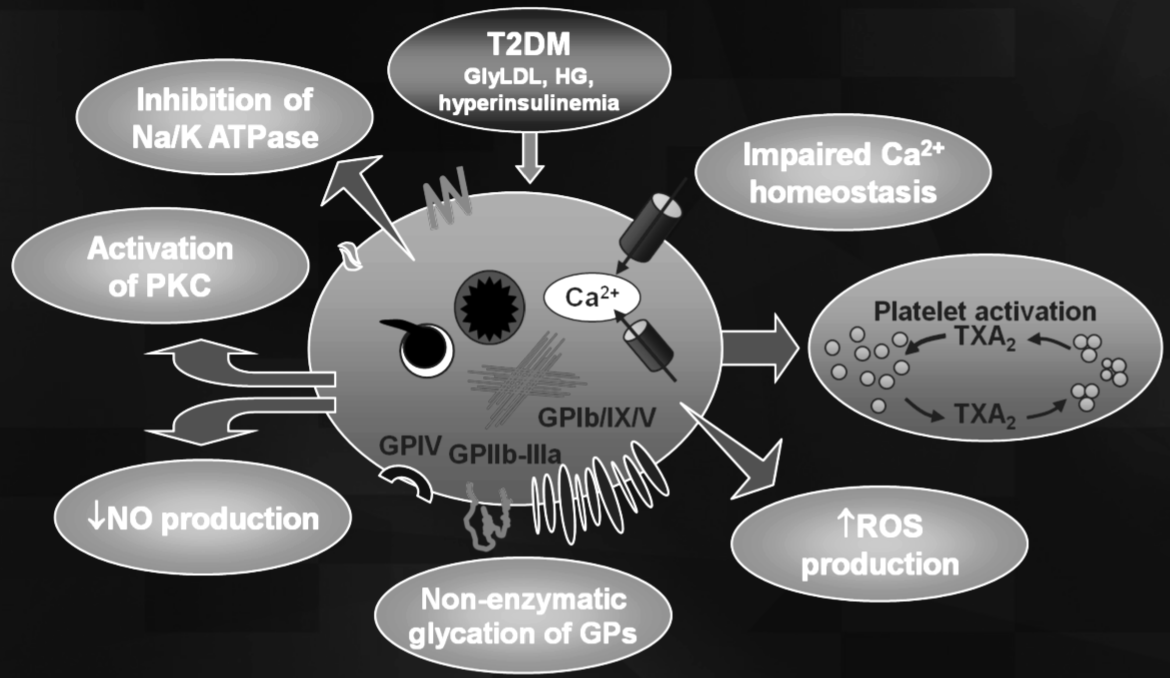
Figure 1: Impact of hyperglycaemia on platelet function.
Non-enzymatic glycation of platelet membrane proteins that decreases membrane fluidity, which may increase platelet adhesion [28],
Osmotic effect of glucose that activates platelet GP IIb/IIIa and P-selectin expression [29],
Activation of protein kinase C, a mediator of platelet activation [30], and
Glycation of circulating Low-Density Lipoproteins (LDL), which increases intracellular calcium concentration and Nitric Oxide (NO) production [31].
It has been shown that challenging healthy non-diabetic subjects’ with24h hyperglycaemia-hyperinsulinemia altered the insulin signalling pathway [32]. Indeed, there is an upregulation on mRNA and protein levels of both glycogen synthase kinase 3Β (GSKΒ3) and tyrosine phosphatase SHP2 as well as tissue factor, while in the meantime there is a downregulation on mRNA for the syntaxin 4-binding protein. High glucose can also increase Calcium influx in platelets is also increased during high glucose levels through enhancement of the PI3K-dependent transient receptor potential channel canonical type 6 [33]. The latter is well-known to be significantly extremely expressed in diabetic platelets. Hyperglycaemia is able to increase the expression and/or activity of Protein Kinase C (PKC) [30], a central kinase in the regulation of platelet activity. hyperglycaemia affects simultaneously the mitochondrial function in platelets. One of the recent components that link hyperglycaemia and mitochondrial dysfunction is the activation of aldose reductase and subsequent ROS production which result to p53 phosphorylation [34] or the stimulation of the PLC?2/PKC/p38aMAPK pathway and the increase in TXA2 production [35]. More recent studies reported that the platelet activation due to hyperglycaemia could be involved into the down regulation of different micro-RNA (miR) such as miR- 223 and miR-146a. It was found that low platelet and plasmamiR-223 and miR-146a expression has been related with an increased risk for ischemic stroke in patients with diabetes mellitus [36]. Many of the harmful effects of glucose have been associated to its metabolite Methylgyloxal (MG). It is known that plasma levels of MG in diabetic patients are higher. Study examining the effect of MGon platelets showed that it increases intracellular Ca2+ levels and activates classical PKCs while inhibiting PI3K/Akt and the Β3-integrinoutsidein signalling. Moreover, in vivo, MG increases thrombus size but reduces its stability in mice [37]. Although most of the effects of MG have been attributed to the formation of Advanced Glycation Endproducts (AGEs) and the subsequent activation of the AGE receptor, studies showed a direct effect of MG on platelets which may contribute to the diabetes-associated platelet hyperaggregability. The expression of surface markers such as P-select in (CD62) and CD63 (a lysosomal glycoprotein) has been shown to be significantly augmented by AGE stimulation [38] suggesting AGE-induced platelet degranulation. Studies done in mice could show that AGEs develop a prothrombotic phenotype via interaction with platelet glycoprotein IV (CD36) [39]. The serum- and glucocorticoid-inducible kinase 1 (SGK1) has been equally suggested mediating AGE-induced platelet hyper activation [40]. In platelets, SGK1 intensify Store-Operated Calcium Entry (SOCE) and so controls several Ca2+-dependent platelet functions such as degranulation, phosphatidylserine exposure, integrin aIIbΒ3 activation, aggregation and thrombus formation. Here, it is worth mentioning that hyperglycaemia also stimulates thrombotic events through plasmatic mechanisms. In particular, hyperglycaemia activates coagulation by raising concentrations of procoagulant factors (e.g. tissue factor, von Willebrand factor) [41] and inhibits fibrinolysis by increasing concentrations of plasminogen activator inhibitor.
Insulin Deficiency and Resistance
The vast majority of DM patients can be grouped to two broad aetiopathogenetic categories. Type-1 DM, which accounts for only 5–10% of cases, results from a cellular-mediated autoimmune destruction of Β-cells of the pancreas, leading to an absolute deficiency of insulin secretion. Type-2 DM, which accounts for around 90–95% of DM individuals, is caused by a combination of resistance to insulin action and an inadequate compensatory insulin secretory response, usually having relative insulin deficiency [16].
Deficient insulin action is the fundamental factor for development of DM and clearly contributes to platelet dysfunction [42]. Both Insulin Receptors (IR) and Insulin-like Growth Factor-1 (IGF-1) receptors are expressed in platelets [43]. Among other effects in platelets, insulin increases surface expression of adenylate cyclaselinked prostacyclin (PGI2) receptor and induces the release of plasminogen activator. IGF-1 is existing in alpha granules of platelets and high levels of its functional receptor are expressed on the platelet surface, which may be involved to the amplification of platelet responses. IGF-1 stimulation of platelets results in dose-dependent phosphorylation of the IGF receptor and in tyrosine phosphorylation of Insulin Receptor Substrate-1 (IRS-1) and IRS-2, stimulating their subsequent binding with the p85 subunit of Phosphoinositide-3 kinase (PI3K), leading to phosphorylation of protein kinase B, which is involved in several cellular responses to insulin and IGF-1, including modulation of platelet reactivity [44].
Several anomalies in insulin-mediated signalling pathways, which can be categorized as IRS-dependent and independent factors, have been proposed to contribute to the vulnerable or ended plateletinhibitory effect detected in patients with insulin resistance [42,45]. Among IRS-dependent factors, insulin resistance causes increased intracellular calcium concentration that leads to augmented platelet degranulation and aggregation [46]. Among IRS-independent pathways contribute to platelet dysfunction due to insulin resistance, the role of reduced platelet sensitivity to NO and PGI2 is remarkable, since both molecules released by the endothelium delay platelet activation; consequently, an impaired response is related with superior platelet reactivity [47]. Further, a recent study found connection between improvement of insulin sensitivity, measured by the Homeostasis Model Assessment (HOMA) of insulin resistance, and changes of platelet function [48].
The significance of insulin resistance in platelet hyperreactivity among patients with DM is underlined by recent studies with thiazolidinediones in which this group of insulin sensitizers has shown a positive effect on platelet function. For instance, rosiglitazone therapy has been associated in some preliminary studies with better sensitivity to NO and partial control of intracellularcalcium concentrations in DM patients [49]. There are controversial data regarding the proposed benefit of insulin-sensitizer therapy over insulin-providing therapy in terms of atherosclerosis progression and cardiovascular outcomes. The PERISCOPE [50] showed that Pioglitazone, an insulin-sensitizer, therapy was related with a significantly lower rate of atherosclerosis progression at 18 months, when compared with glimepiride, an insulin secretagogue, treatment. The result also has been reported in SYMPHONY substudy [51]. Although these studies highlighted the central role of insulin resistance in progression of atherothrombotic events in DM patients, these outcomes have not been established in the later published APPROACH trial [52]. In this study, there was no significant difference between the group treated with thiazolinedione rosiglitazone and the group treated with glipizide, an insulin secretagogue, in patients with type-2 DM and CAD [52].
Related Metabolic Conditions
Several metabolic conditions connected with type-2 DM may are responsible for the increased platelet reactivity saw in these patients, including obesity, dyslipidaemia and increased systemic inflammation.
Obesity is a common characteristic in patients with type-2 DM. First of all obesity itself may be associated with insulin resistance, which has relevant effects on platelet reactivity as described above. Nevertheless, other factors present in obese patients may be the reason for platelet dysfunction, which include:
Overall, these metabolic abnormalities observed to obese patients eventually lead to increased platelet reactivity [53,54]. Russo et al. recently reported that weight loss returned the sensitivity to NO and PGI2 and reduced platelet activation [48]. In any case, response to anti platelet drugs, mainly to clopidogrel, is impaired in persons with high body mass index [55].
Dyslipidaemia is also common among the type-2 DM patients. It is typically expressed by elevated triglycerides and low High- Density Lipoprotein (HDL) cholesterol. Hypertriglyceridemia is a characteristic manifestation that is known to enhance platelet activation [56]. Triglyceride-rich Very-Low-Density Lipoprotein (VLDL) particles also influences the coagulation cascade and harmsthe fibrinolysis, thus predisposing to atherothrombosis [57]. The recommended mechanism to elucidate the effects of triglycerides on platelet function is the apoE content of VLDL particles and their contact with the platelet LDL receptor [58]. Moreover, the thrombotic properties of LDL seem to be rather related to its oxidation. The oxidized-LDL can directly interact with platelets specific receptors such as the lectin-likeoxidized LDL receptor-1 [59] or the CD36 [60,61] and contribute to platelet’s activation. Not only ox-LDL activates platelets but activated platelets are also known to be able to produce ox-LDL upholding the vicious cycle. Low levels of HDL are related with endothelial dysfunction, which increases the atherothrombotic risk in DM patients. On the other hand, intravenous administration of reconstituted HDL rapidly controls the endothelial function by increasing NO bioavailability in patients with hypercholesterolaemia [62]. Similarly, Calkin et al reported that reconstituted HDL caused a reduction of platelet aggregability in DM patients by flowing out cholesterol from platelets [63].
DM is a medical condition characterized by systemic inflammation. Indeed, DM patients have higher concentrations of inflammatory, platelet activation and coagulation markers, such as soluble CD40 lig and, soluble P-select in, interleukin-6, and tissue factor, compared with healthy subjects [64]. Furthermore, inflammation modifies the expression of Fc gamma RIIA receptor, which is higher in platelets of DM patients and helps to platelet activation [65].
Additional Cellular Abnormalities
Several studies have proved that other platelet anomalies common in DM patients may cause their hyperreactivity status, including dysregulation of calcium metabolism, oxidative stress, upregulation of P2Y12 signalling pathway and accelerated platelet turnover.
Platelets from patients with DM have increased intracellular calcium levels due to impaired modulation of calcium metabolism. The factors involved in calcium signalling abnormalities in DM patients are not fully clarified. Mechanisms that have been proposed to impair calcium homeostasis and, consequently, in platelet hyperreactivity are: a) disproportionate influx of calcium through the sodium/calcium exchanger [66], b) alterations in the activity of calcium ATPases [67], c) reduced sensitivity to insulin, which decreases sarcoplasmic endoplasmic reticulum calcium-ATPase (SERCA-2) [42,49], and d) increased oxidative stress, which induces calcium signalling due to increased formation of superoxide anions and reduced nitric oxide production [68]. The final result of this altered calcium homeostasis is an augmented concentration of cytosolic calcium, which leads to enhanced platelet reactivity and aggregation [42].
Platelet function is influenced by the presence of reactive oxygen species as well as by the regulation of platelet or vascular redox status, and the platelets may become prothrombotic [69]. DM is related with oxidative stress, mainly with an overproduction of reactive oxygen and nitrogen species, as well as reduced platelet antioxidant levels [70]. The platelet activation is increased due to excessive generation of potent oxidants, such as superoxide anions and hydrogen peroxide [69,71] An increase in reactive oxygen species along with chronic hyperglycaemia augments production of advanced glycation end products (AGEs) [72]. AGEs may developatherosclerotic complications by activation of the receptor for AGEs (RAGE) [73] and by enhancing platelet aggregation through the serotonin receptor [74]. Further, endothelial function is impaired also by oxidative stress through a reduction in production of NO and PGI2 [75]. This is of particular interest in patients with DM since their platelets have diminished sensitivity to the actions of these vasodilator molecules produced by the endothelium [47]. Recently it has been showed that high blood glucose levels trigger neutrophil release of S100 calciumbinding protein A8/A9 (S100A8/A9), which binds to the receptor for advanced glycation end products (RAGE) on Kupffer cells, ultimately leading to increased Thrombopoietin (TPO) production in the liver. TPO causes megakaryocyte proliferation and consequently thrombocytosis [76].
The platelet Adenosine Diphosphate (ADP) P2Y12 receptor signalling pathway has been suggested to be upregulated in diabetic platelets, in particular those with type-2 DM [77]. This suppresses cAMP concentration and, in addition to a lower responsiveness to insulin, leads to increased adhesion, aggregation, and procoagulant activity [78]. Another abnormality in platelet surface molecules is the increased expression of surface proteins such as P-selectin and glycoproteins Ib and IIb/IIIa, which are integrins that mediate adhesion [53].
An accelerated platelet turnover represented by the presence of a higher number of reticulated platelets has been observed in patients with DM. Reticulated platelets are larger and more sensitive, resulting in platelet hyperreactivity and lower response to antiplatelet therapies including aspirin and clopidogrel [79,80]. In addition, platelets of DM patients are larger, and thus hyperreactive, as platelet size correlates with greater platelet reactivity measured by aggregation and total release of granular content.
Increased Apoptosis
Although being anucleated, there is evidence indicating that platelets have the required machinery to undergo apoptosis [81]. Among other, calpain seems to play an important role in platelet apoptosis [82]. Indeed, caspase activation during platelet apoptosis seems to be downstream of calpain activation. The increased calpain activation observed in DM platelets suggests that diabeticplatelets may be more susceptible to apoptosis. Several factors have been reported to induce platelet apoptosis including the diabetes-associated oxidative stress which is an important stimulus for inducing mitochondrial damage [83]. Mitochondria not only are targeted by the oxidative stress but may also amplify the reaction to oxidative stress due to ROS generation [83,84]. The mitochondrial function of diabetic platelets is altered due to increased ATP content and decreased mitochondrial membrane potential [85]. Another mechanism involved in platelet apoptosis is the development of Endoplasmic Reticulum (ER) stress due to overproduction of AGEs [86] and homocysteine [87].
One of the consequences of platelet activation and apoptosis is the generation of intact membrane vesicles known as microparticles. The formation of platelet-derived microparticles (PMPs) is known to be calcium and calpain dependent. Although PMPs are involved in haemostasis due to their procoagulant properties, elevated levels of PMPs in blood from diabetic patients has been suggested to take part in the increased vascular complications in diabetes [88,89].
Increased Mean Platelet Volume
As mentioned above platelet reactivity and size have been shown to directly correlate. Indeed, young and large platelets exhibit higher activity than old and small ones. The Mean Platelet Volume (MPV) is an indicator of the average size of platelets which has been largely used to investigate the relationship between platelet size and activity. There is evidence that MPV is significantly increased in diabetic patients and that it directly associates with glycaemic control and obesity [90].
Hyporesponsiveness to Anti-Platelet Therapy
One other feature of diabetic platelets is their hyporesponsiveness to anti-platelet therapy. Indeed, there is evidence that anti-platelet therapy is significantly less effective in diabetic patients when compared with patients without diabetes [13]. Diabetic patients are refractory to the anti-platelet effect of aspirin, a phenomenon called “aspirin resistance” [91]. Aspirin or salicylic acid acetylates and irreversibly inhibits cyclooxygenase thus inhibiting the TXA2 formation. Although aspirin resistance is seen in the majority of diabetic patients, the exact molecular mechanism is still blurred. One of the mechanisms suggested to contribute to aspirin-resistance is the increased glycation of platelet proteins which may alter the acetylation process [92]. Many in vitro studies have also shown a direct connection between hyperglycaemia and aspirin resistance. Indeed, high glucose can directly reduce the anti aggregating effect of aspirin by inhibiting the aspirin-induced activation of the NO/ cGMP/PKG pathway without affecting the aspirin induced inhibition of TXA2 synthesis [93]. It also has been suggested that lactic acid might be the mediator of the glucose-induced inhibition of the aspirin effect in platelets [94]. More recently, non-HDL cholesterol has also been reported to be an independent risk factor for aspirin resistance in patients with type 2 diabetes [95].
Diabetes is also known to be associated to a reduced responsiveness of platelets to the P2Y12ADP receptor antagonist clopidogrel [10,96]. Although not directly investigated in diabetic patients, upstream of ADP receptor levels may contribute to clopidogrel resistance.
Conclusions
The fact that the diabetic environment can modulate platelet function in several ways describes the failure of glycaemic control alone to reduce the risk of atherothrombotic events in diabetic patients. Certainly, the increased platelet hyper reactivity is the outcome of complicated inter-regulated mechanisms (Figure 2). Additionally, bearing in mind that diabetic platelets are resistant to most anti-platelet therapy, there is a need of new therapeutical strategies to improve platelet function in diabetes. Definitely, controlling both glycemia and dyslipidaemia may improve the effects of antiplatelet therapy. The understanding of the molecular mechanisms leading to increased platelet reactivity in patients with DM may set the basis for targeted antiplatelet treatment strategies in this high-risk cohort. Thus, the facts that calpain plays an important role in platelet activation and that calpain activity is elevated in diabetic platelets, makes it attractive to propose the Ca2+-activated proteases as a promising therapeutic target to prevent thrombotic complications in diabetic patients.
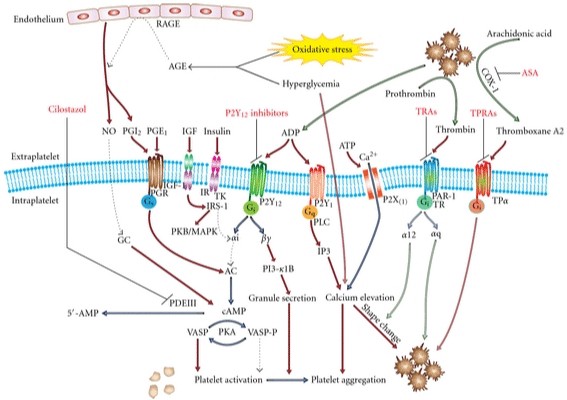
Figure 2: Pathways involved in platelet hyper reactivity in DM patients and therapeutic targets.
References
- Webster MW, Scott RS. What cardiologists need to know about diabetes. The Lancet. 1997; 350: S23-S28.
- S Haffner, S Lehto, T Rönnemaa, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998; 339: 229-34.
- T Lüscher, M Creager, J Beckman, F Cosentino. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part II. Circulation. 2003; 108: 1655-61.
- M Roffi, D P Chew, D Mukherjee, D L Bhatt, J A White, et al. Platelet glycoprotein IIb/IIIa inhibitors reduce mortality in diabetic patients with non- ST-segment-elevation acute coronary syndromes. Circulation. 2001; 104: 2767-71.
- Gerard X Brogan, Eric D Peterson, Jyotsna Mulgund, Deepak L Bhatt, E Magnus Ohman, et al. Treatment disparities in the care of patients with and without diabetes presenting with non-ST-segment elevation acute coronary syndromes. Diabetes Care. 2006; 29: 9-14.
- Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and Vascular Disease: Pathophysiology, Clinical Consequences, and Medical Therapy: Part I. Circulation: Journal of the American Heart Association. 2003; 108: 1527-1532.
- Osende JI, Badimon JJ, Fuster V, Herson P, Rabito P, Vidhun R, et al. Blood thrombogenicity in type 2 diabetes mellitus patients is associated with glycemic control. Journal of the American College of Cardiology. 2001; 38: 1307-1312.
- G Davì, C Patrono. Platelet activation and atherothrombosis. N Engl J Med. 2007; 357: 2482-94.
- B Stratmann, D Tschoepe. Pathobiology and cell interactions of platelets in diabetes. Diab Vasc Dis Res. 2005; 2: 16-23.
- Angiolillo DJ, Fernandez-Ortiz A, Bernardo E, Ramírez C, Sabaté M, Jimenez-Quevedo P, et al. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes. 2005; 54: 2430-2435.
- Vinik AI, Erbas T, Park TS, Nolan R, Pittenger GL. Platelet dysfunction in type 2 diabetes. Diabetes care. 2001; 24: 1476-1485.
- P Ferroni, S Basili, A Falco, G Davì, Platelet activation in type 2 diabetes mellitus. J Thromb Haemost. 2004; 2: 1282-91.
- Angiolillo DJ. Antiplatelet Therapy in Diabetes: Efficacy and Limitations of Current Treatment Strategies and Future Directions. Diabetes Care. 2009; 32: 531-540.
- A Natarajan, A Zaman, S. Marshall. Platelet hyperactivity in type 2 diabetes: role of antiplatelet agents. Diab Vasc Dis Res. 2008; 5: 138-44.
- J Ferreiro, D Angiolillo. Diabetes and antiplatelet therapy in acute coronary syndrome. Circulation. 2011; 123: 789-813.
- DA American. Classification and Diagnosis of Diabetes. Diabetes Care. 2017; 40: S11-S24.
- D Schneider. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes Care. 2009; 32: 525-7.
- Lemkes BA, Hermanides J, Devries JH, Holleman F, Meijers JCM, Hoekstra JBL. Hyperglycemia: a prothrombotic factor?. Journal of Thrombosis and Haemostasis. 2010; 8: 1663-1669.
- N Eibl, W Krugluger, G Streit, K Schrattbauer, P Hopmeier, G Schernthaner. Improved metabolic control decreases platelet activation markers in patients with type-2 diabetes. Eur J Clin Invest. 2004; 34: 205-9.
- Gresele P, Guglielmini G, Angelis MD, Ciferri S, Ciofetta M, Falcinelli E, et al. Acute, short-term hyperglycemia enhances shear stress-induced platelet activation in patients with type II diabetes mellitus. Journal of the American College of Cardiology. 2003; 41: 1013-1020.
- M Yngen, C G Ostenson, N Li, P Hjemdahl, N H Wallén. Acute hyperglycemia increases soluble P-selectin in male patients with mild diabetes mellitus. Blood Coagul Fibrinolysis. 2001; 12: 109-16.
- Undas A, Wiek I, Stêpien E, Zmudka K, Tracz W. Hyperglycemia Is Associated With Enhanced Thrombin Formation, Platelet Activation, and Fibrin Clot Resistance to Lysis in Patients With Acute Coronary Syndrome. Diabetes Care. 2008; 31: 1590-1595.
- M Yngen, A Norhammar, P Hjemdahl, N Wallén. Effects of improved metabolic control on platelet reactivity in patients with type 2 diabetes mellitus following coronary angioplasty. Diab Vasc Dis Res. 2006; 3: 52-6.
- Malmberg K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. BMJ. 1997; 314: 1512-1512.
- K Malmberg, L Rydén, H Wedel, K Birkeland, A Bootsma, et al. Intense metabolic control by means of insulin in patients with diabetes mellitus and acute myocardial infarction (DIGAMI 2): effects on mortality and morbidity. Eur Heart J. 2005; 26: 650-61.
- Hertzel C Gerstein, Michael E Miller, Robert P Byington, David C Goff Jr, J Thomas Bigger, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008; 358: 2545-59.
- Simon Finfer, Dean R Chittock, Steve Yu-Shuo Su, Deborah Blair, Denise Foster, et al. Intensive versus conventional glucose control in critically ill patients. N Engl J Med. 2009; 360: 1283-97.
- Watala C, Golanski J, Boncler MA, Pietrucha T, Gwozdzinski K. Membrane lipid fluidity of blood platelets: a common denominator that underlies the opposing actions of various agents that affect platelet activation in whole blood. Platelets. 1998; 9: 315-327.
- Keating FK, Sobel BE, Schneider DJ. Effects of increased concentrations of glucose on platelet reactivity in healthy subjects and in patients with and without diabetes mellitus. The American journal of cardiology. 2003; 92: 1362-1365.
- R Assert, G Scherk, A Bumbure, et al. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia. 2001; 44: 188-95.
- Ferretti G, Rabini RA, Bacchetti T, Vignini A, Salvolini E, Ravaglia F, et al. Glycated low density lipoproteins modify platelet properties: a compositional and functional study. The Journal of clinical endocrinology and metabolism. 2002; 87: 2180-2184.
- A Rao, R Freishtat, G Jalagadugula, Singh A, Mao G, et al. Alterations in insulin-signaling and coagulation pathways in platelets during hyperglycemiahyperinsulinemia in healthy non-diabetic subject.," Thromb Res. 2014; 134: 704-10.
- D Liu, A Maier, A Scholze, Rauch U, Boltzen U, et al. High glucose enhances transient receptor potential channel canonical type 6-dependent calcium influx in human platelets via phosphatidylinositol 3-kinase-dependent pathway. Arterioscler Thromb Vasc Biol. 2008; 28: 746-51.
- Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, et al. Aldose Reductase– Mediated Phosphorylation of p53 Leads to Mitochondrial Dysfunction and Damage in Diabetic Platelets. Circulation. 2014; 129: 1598-1609.
- W Tang, J Stitham, S Gleim, Di Febbo C, Porreca E, et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J Clin Invest. 2011; 121: 4462-76.
- Duan X, Zhan Q, Song B, Zeng S, Zhou J, Long Y, et al. Detection of platelet microRNA expression in patients with diabetes mellitus with or without ischemic stroke. Journal of diabetes and its complications. 2014; 28: 705- 710.
- K Hadas, V Randriamboavonjy, A Elgheznawy, Mann A, Fleming I. Methylglyoxal induces platelet hyperaggregation and reduces thrombus stability by activating PKC and inhibiting PI3K/Akt pathway. PLoS One. 2013; 8: e74401.
- Gawlowski T, Stratmann B, Ruetter R, Buenting CE, Menart B, Weiss J, et al. Advanced glycation end products strongly activate platelets. European Journal of Nutrition. 2009; 48: 475-481.
- Zhu W, Li W, Silverstein RL. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood. 2012; 119: 6136-6144.
- Lang F, Gawaz M, Borst O. The serum & glucocorticoid-inducible kinase in the regulation of platelet function. Acta Physiologica. 2015; 213: 181-190.
- G Boden, A Rao. Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr Diab Rep. 2007; 7: 223-7.
- Randriamboavonjy V, Fleming I. Insulin, Insulin Resistance, and Platelet Signaling in Diabetes. Diabetes Care. 2009; 32: 528-530.
- Hunter RW, Hers I. Insulin/IGF-1 hybrid receptor expression on human platelets: consequences for the effect of insulin on platelet function. Journal of Thrombosis and Haemostasis. 2009; 7: 2123-2130.
- I Hers. Insulin-like growth factor-1 potentiates platelet activation via the IRS/ PI3Kalpha pathway. Blood. 2007; 110: 4243-52.
- Westerbacka J, Yki-Järvinen H, Turpeinen A, Rissanen A, Vehkavaara S, Syrjälä M, et al. Inhibition of Platelet-Collagen Interaction An In Vivo Action of Insulin Abolished by Insulin Resistance in Obesity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002; 22: 167-172.
- Ferreira IA, Eybrechts KL, Mocking AIM, Kroner C, Akkerman JN. IRS-1 Mediates Inhibition of Ca2+ Mobilization by Insulin via the Inhibitory G-protein Gi*. Journal of Biological Chemistry. 2004; 279: 3254-3264.
- G Anfossi, E M Mularoni, S Burzacca, M C Ponziani, P Massucco. Platelet resistance to nitrates in obesity and obese NIDDM, and normal platelet sensitivity to both insulin and nitrates in lean NIDDM. Diabetes Care. 1998; 21: 121-6.
- Russo I, Traversa M, Bonomo K, Salve A, Mattiello L, Mese P, et al. In Central Obesity, Weight Loss Restores Platelet Sensitivity to Nitric Oxide and Prostacyclin. Obesity. 2010; 18: 788-797.
- V Randriamboavonjy, F Pistrosch, B Bölck, Schwinger RHG, Dixit M, et al. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation. 2008; 117: 52-60, 2008.
- Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA. 2008; 299: 1561.
- McGuire DK, Newby LK, Bhapkar MV, Moliterno DJ, Hochman JS, Klein WW, et al. Association of diabetes mellitus and glycemic control strategies with clinical outcomes after acute coronary syndromes. American heart journal. 2004; 147: 246-252.
- Hertzel C Gerstein, Robert E Ratner, Christopher P Cannon, Patrick W Serruys, Héctor M García-García, et al. Effect of rosiglitazone on progression of coronary atherosclerosis in patients with type 2 diabetes mellitus and coronary artery disease: the assessment on the prevention of progression by rosiglitazone on atherosclerosis in diabetes patients with cardiov. Circulation. 2010; 121: 1176-87.
- D Schneider, R Hardison, N Lopes, Sobel EB, Brooks MM, et al. Association between increased platelet P-selectin expression and obesity in patients with type 2 diabetes: a BARI 2D (Bypass Angioplasty Revascularization Investigation 2 Diabetes) substudy. Diabetes Care. 2009; 32: 944-9.
- T Murakami, H Horigome, K Tanaka, Nakata Y, Ohkawara K, et al Impact of weight reduction on production of platelet-derived microparticles and fibrinolytic parameters in obesity. Thromb Res. 2007; 119: 45-53.
- Sibbing D, Beckerath OV, Schömig A, Kastrati A, Beckerath NV. Impact of body mass index on platelet aggregation after administration of a high loading dose of 600 mg of clopidogrel before percutaneous coronary intervention. The American journal of cardiology. 2007; 100: 203-205.
- Man FHD, Nieuwland R, Laarse AVD, Romijn F, Smelt AH, Leuven JAG, et al. Activated platelets in patients with severe hypertriglyceridemia: effects of triglyceride-lowering therapy. Atherosclerosis. 2000; 152: 407-414.
- Olufadi R, Byrne CD. Effects of VLDL and Remnant Particles on Platelets. Pathophysiology of Haemostasis and Thrombosis. 2006; 35: 281-291.
- Pedreño J, Hurt-Camejo E, Wiklund O, Badimón L, Masana L. Platelet function in patients with familial hypertriglyceridemia: evidence that platelet reactivity is modulated by apolipoprotein E content of very-low-density lipoprotein particles. Metabolism: clinical and experimental. 2000; 49: 942- 949.
- M Chen, M Kakutani, T Naruko, Ueda M, Narumiya S, et al. Activationdependent surface expression of LOX-1 in human platelets. Biochem Biophys Res Commun. 2001; 282: 153-8.
- K Chen, M Febbraio, W Li, R Silverstein. A specific CD36-dependent signaling pathway is required for platelet activation by oxidized low-density lipoprotein. Circ Res. 2008; 102: 1512-9.
- E Podrez, T Byzova, M Febbraio, Salomon RG, Ma Y, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007; 13: 1086-95.
- Spieker LE, Sudano I, Hürlimann D, Lerch PG, Lang MG, Binggeli C, et al. High- Density Lipoprotein Restores Endothelial Function in Hypercholesterolemic Men. Circulation: Journal of the American Heart Association. 2002; 105: 1399-1402.
- Calkin AC, Drew BG, Ono A, Duffy SJ, Gordon MV, Schoenwaelder SM, et al. Reconstituted High-Density Lipoprotein Attenuates Platelet Function in Individuals With Type 2 Diabetes Mellitus by Promoting Cholesterol Efflux. Circulation. 2009; 120: 2095-2104.
- Lim HS, Blann AD, Lip GYH. Soluble CD40 Ligand, Soluble P-Selectin, Interleukin-6, and Tissue Factor in Diabetes Mellitus: Relationships to Cardiovascular Disease and Risk Factor Intervention. Circulation: Journal of the American Heart Association. 2004; 109: 2524-2528.
- Calverley DC, Hacker MR, Loda KA, Brass E, Buchanan TA, Tsao-Wei DD, et al. Increased platelet Fc receptor expression as a potential contributing cause of platelet hypersensitivity to collagen in diabetes mellitus. British Journal of Haematology. 2003; 121: 139-142.
- Li Y, Woo V, Bose R. Platelet hyperactivity and abnormal Ca(2+) homeostasis in diabetes mellitus. American journal of physiology. Heart and circulatory physiology. 2001; 280: H1480-H1489.
- M Balasubramanyam, R Balaji, B Subashini, V Mohan. Evidence for mechanistic alterations of Ca2+ homeostasis in Type 2 diabetes mellitus. Int J Exp Diabetes Res. 2001; 1: 275-87, 2001.
- Schaeffer G, Wascher TC, Kostner GM, Graier WF. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia. 1999; 42: 167-176.
- J Freedman. Oxidative stress and platelets. Arterioscler Thromb Vasc Biol. 2008; 3: s11-6.
- Jardín I, Redondo PC, Salido GM, Pariente JA, Rosado JA. Endogenously generated reactive oxygen species reduce PMCA activity in platelets from patients with non-insulin-dependent diabetes mellitus. Platelets. 2006; 17: 283-288.
- P Redondo, I Jardin, J Hernández-Cruz, Pariente JA, Salido GM, et al. Hydrogen peroxide and peroxynitrite enhance Ca2+ mobilization and aggregation in platelets from type 2 diabetic patients. Biochem Biophys Res Commun. 2005; 333: 794-802.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000; 404: 787-790.
- A Schmidt, S Yan, J Wautier, D Stern. Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res. 1999; 84: 489-97.
- Y Hasegawa, A Suehiro, S Higasa and e. al, Enhancing effect of advanced glycation end products on serotonin-induced platelet aggregation in patients with diabetes mellitus. Thromb Res. 2002; 107: 319-23.
- Schäfer A, Bauersachs J. Endothelial dysfunction, impaired endogenous platelet inhibition and platelet activation in diabetes and atherosclerosis. Current vascular pharmacology. 2008; 6: 52-60.
- Lee RH, Bergmeier W. Sugar makes neutrophils RAGE: linking diabetesassociated hyperglycemia to thrombocytosis and platelet reactivity. The Journal of clinical investigation. 2017; 127: 2040-2043.
- Matsuno H, Tokuda H, Ishisaki A, Zhou Y, Kitajima Y, Kozawa O. P2Y12 receptors play a significant role in the development of platelet microaggregation in patients with diabetes. The Journal of clinical endocrinology and metabolism. 2005; 90: 920-927.
- Ferreira IA, Mocking AIM, Feijge MAH, Gorter G, Haeften TWV, Heemskerk JWM, et al. Platelet Inhibition by Insulin Is Absent in Type 2 Diabetes Mellitus. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006; 26: 417-422.
- Guthikonda S, Lev EI, Patel R, DeLao T, Bergeron AL, Dong J, et al. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. Journal of Thrombosis and Haemostasis. 2007; 5: 490-496.
- S Guthikonda, C Alviar, M Vaduganathan, Arikan M, Tellez A, et al. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol. 2008; 52: 743-9.
- Leytin V. Apoptosis in the anucleate platelet. Blood Rev. 2012; 26: 51-63.
- W Zhang, J Liu, R Sun, Zhao L, Du J, et al. Calpain activator dibucaine induces platelet apoptosis. Int J Mol Sci. 2011 12: 2125-37.
- Garcia-Souza LF, Oliveira MF. Mitochondria: biological roles in platelet physiology and pathology. The international journal of biochemistry & cell biology. 2014; 50: 156-160.
- Pietraforte D, Vona R, Marchesi A, Jacobis ITD, Villani A, Principe DD, et al. Redox control of platelet functions in physiology and pathophysiology. Antioxidants & redox signaling. 2014;21(1):177-193. doi:10.1089/ ars.2013.5532
- Ran J, Guo X, Li Q, Mei G, Lao G. Platelets of type 2 diabetic patients are characterized by high ATP content and low mitochondrial membrane potential. Platelets. 2009; 20: 588-593.
- Vera RH, Vilahur G, Ferrer-Lorente R, Peña E, Badimon L. Platelets Derived From the Bone Marrow of Diabetic Animals Show Dysregulated Endoplasmic Reticulum Stress Proteins That Contribute to Increased Thrombosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012; 32: 2141-2148.
- Zbidi H, Redondo PC, López JJ, Bartegi A, Salido GM, Rosado JA. Homocysteine induces caspase activation by endoplasmic reticulum stress in platelets from type 2 diabetics and healthy donors. Thrombosis and haemostasis. 2010; 103: 1022-1032.
- X Zhang, S McGeoch, A Johnstone, G Holtrop, Sneddon AA, et al. Plateletderived microparticle count and surface molecule expression differ between subjects with and without type 2 diabetes, independently of obesity status. J Thromb Thrombolysis. 2014; 37: 455-63.
- Salem MAEK, Adly AAM, Ismail EAR, Darwish YW, Kamel HA. Platelets microparticles as a link between micro- and macro-angiopathy in young patients with type 1 diabetes. Platelets. 2015; 26: 682-688.
- T Kodiatte, U Manikyam, S Rao, Jagadish TM, Reddy M, et al. Mean platelet volume in Type 2 diabetes mellitus. J Lab Physicians. 2012; 4: 5-9.
- Ajjan R, Storey RF, Grant PJ. Aspirin resistance and diabetes mellitus. Diabetologia. 2007; 51: 385-390.
- Watala C, Pluta J, Golanski J, Rozalski M, Czyz M, Trojanowski Z, et al. Increased protein glycation in diabetes mellitus is associated with decreased aspirin-mediated protein acetylation and reduced sensitivity of blood platelets to aspirin. Journal of Molecular Medicine. 2004; 83: 148-158.
- Russo I, Viretto M, Barale C, Mattiello L, Doronzo G, Pagliarino A, et al. High Glucose Inhibits the Aspirin-Induced Activation of the Nitric Oxide/cGMP/ cGMP-Dependent Protein Kinase Pathway and Does Not Affect the Aspirin- Induced Inhibition of Thromboxane Synthesis in Human Platelets. Diabetes. 2012; 61: 2913-2921.
- Kobzar G, Mardla V, Samel N. Short-term exposure of platelets to glucose impairs inhibition of platelet aggregation by cyclooxygenase inhibitors. Platelets. 2011; 22: 338-344.
- Kim JD, Park C, Ahn KJ, Cho JH, Choi KM, Kang JG, et al. Non-HDL cholesterol is an independent risk factor for aspirin resistance in obese patients with type 2 diabetes. Atherosclerosis. 2014; 234: 146-151.
- T Geisler, N Anders, M Paterok, Langer H, Stellos K, et al. Platelet response to clopidogrel is attenuated in diabetic patients undergoing coronary stent implantation. Diabetes Care. 2007; 30: 372-4.