
Research Article
Austin Food Sci. 2016; 1(1): 1001.
Biosynthesis of Phosphatidyl Vitamin B6 by Phospholipase D Catalyzed Transphosphatidylation
Guo Z* and Xu X
Department of Engineering, Aarhus University, Denmark
*Corresponding author: Zheng Guo, Department of Engineering, Aarhus University, Denmark
Received: January 18, 2016; Accepted: January 30, 2016; Published: February 05, 2016
Abstract
Commercially available vitamin B6 (VB6) is the hydrochloric salt of pyridoxol,
which is easily excreted with sweat and urea, therefore its bio-utilization rate is
very low. PC (Phosphatidylcholine)-modified VB6 may change its transportation
route, increase the membrane-affinity of VB6 and promote the assimilation
of VB6. Phospholipase D (PLD) was thus used in this work to catalyze head
group exchange of PC with pyridoxol yielding PC-VB6 derivative. The structure
of the synthetic product was characterized by 1H &
Keywords: Pyridoxine (vitamin B6); Phosphatidylcholine (PC); Phospholipase D (PLD); Transphosphatidylation; Phospholipids
Introduction
Being the precursor of pyridoxal phosphate, Vitamin B6 functions as a highly versatile coenzyme in over 100 enzymatic reactions involved in the metabolism of amino acid, carbohydrates, neurotransmitters, and lipids, etc. [1,2]. Clinical evidence has also shown the physiological importance of VB6 in using as a glucocorticoid antagonist, alleviating menstrual irregularities and reducing weight gain with Depo-Provera, etc. [3-5]. There also is growing evidence the high levels of VB6 could suppress growth of cancer cells either in vivo or in vitro [6,7], which represents a new light to view the functionality of VB6. In practice, VB6 has been widely used as pharmaceutical, food supplement and antioxidant in cosmetic formulations [8]. Among three vitamers (pyridoxine, pyridoxamine and pyridoxal), pyridoxine is the main dietary and therapeutic form and usually administered as hydrochlorate. However, water-soluble pyridoxine hydrochlorate generally results in lower biological utilization because of a larger proportion of which excreted with sweat and urine. Lipophilic modification of water-soluble drugs has proved to be an efficient approach to modulate the properties of drugs and simultaneously ensure the cellular availability [9,10]. Because these lipophilic derivatives are supposed to exhibit long half-life but also to be easily distributed to hydrophobic microenvironment of target tissues, where the active ingredients are released by enzymatic interaction [11]. However, after pioneering work of Bbaldessari, et al. [12] who conduct lipase-catalysed esterification of VB6 with fatty acids, few reports concerning Vitamin B6 modification have been published so far. As the integral components of biomembranes, phospholipids not only play ubiquitous and fundamental function for life, but also provide a natural carrier for the delivery and modification of bioactive compounds [11,13,14]. Due to the structural similarity, phosphatidyl derivants are supposed to possess a high affinity to biomembranes and an easy accessibility to cells [13,14]. Phospholipase D (PLD, EC 3.1.4.4) catalyses the cleavage of the terminal phophodiester bond of glycerophopholipids through exchange of head group with water or various acceptor alcohols [15-17]. Thus, besides the hydrolysis resulting in phosphatidic acid, PLD can also serve as an efficient tool to catalyze the transfer of phosphatidyl moiety to the compounds with reactable hydroxyl groups, resulting in novel phospholipids [11,15- 18]. A great number of compounds with physiological or therapeutic activities, such as nucleosides [19], ascorbic acid [20], arbutin and kojic acid [21], have been investigated for transphosphatidylation. These modified products generally showed improved stability and beneficial usability. Structurally, VB6 is a polyfunctional derivative of pyridine, with 2 hydroxymethyl and 1 pheno-hydroxyl groups located at 4-, 5- and 3-position of pyridine ring, respectively (Figure 1). Therefore, it is theoretically feasible to prepare VB6 phosphatidyl derivants with catalysis of PLD. This novel VB6 derivants endow VB6 with different properties, which might help to explore innovative applications of this traditional drug.
To this end, this work presented a systematic study of PLDcatalyzed phosphatidylation of pyridoxine in two-phase system. The structure of the yield product was identified by GC-MS, H1 NMR and C13 NMR analysis. To establish an efficient protocol, reaction conditions involving solvent, enzyme efficiency, temperature and pH etc was optimized.
Materials and Methods
Materials
Phospholipase D (E.C. 3.1.4.4) Type VII from Streptomyces sp. (1550 units/mg solid) and 1,2-Dipalmitoyl-rac-glycero-3- Phosphocholine hydrate (DPPC) were purchased from Sigma- Aldrich (St. Louis, MO). Soybean phosphatidylcholine (with a minimum content 93%) was obtained from Degussa Texturant Systems Deutschland GmbH & Co.KG (Hamburg, Germany). The fatty acid composition (mol %) of soybean PC was C16:0, 13.7; C18: 0, 3.6; C18: 1, 9.5; C18: 2, 66.0; and C18: 3, 7.2. Pyridoxine (free base) was from Sigma-Aldrich Denmark A/S (Broendby, Denmark). Other chemicals and reagents were all of analytical grade and used as received.
Preparation of DPPC-Pyridoxine
10 m.mol pyridoxine was dissolved in 20 mL of 100 mM pH 5.6 sodium acetate buffer containing 40 mM Ca2+. The resulting solution was mixed with 20 mL dichloromethane of 1 mmol DPPC (0.734g). The reaction was initiated by the addition of 50 units of PLD dissolved in pH 5.6 NaOAc buffer (One unit is defined as the activity to liberate 1.0 μmole of choline from L-α-phosphatidylcholine (egg yolk) per hr at pH 5.6 at 30oC). The incubation was conducted at 30oC for 24 hr with magnetic stirring. At the end of the reaction, the organic layer was collected and washed 3 times with deionized water. The organic layer was then dried over anhydrous Na2SO4 and concentrated, and chromatographed on a silica gel column. The first elute with CHCl3- MeOH (3/1, v/v) yielded 637 mg DPPC-VB6. The elute with CHCl3- MeOH-H2O (10/5/1, v/v/v) gave 78.9 mg of recovered DPPC with little impurity of dipalmitoyl-3-phosphatidic acid salt.
Spectral analysis and structural identification
APCI LC/MS: The products and the PL standard mixture were separated on a silica column (l = 15 cm, i.d. = 4.6 mm, particle size = 5 μm, Phenomenex). The column was fitted into an HP 1100 Series LC/MSD system, consisting of a quaternary pump, a vacuum degasser, an autosampler, a diode array detector, and an MS detector (Hewlett-Packard, Waldbronn, Germany). A binary solvent system of chloroform/methanol/ ammonium acetate (90/10/0.5) and chloroform/methanol/water/ammonium acetate (60/35/5/0.5) was used. API-ES was used in the negative mode. The capillary voltage was 4000V, and the drying gas was 350oC and 10 L/min, nebulizer gas pressure was 25 psi. The heated nitrogen drying gas temperature and flow rate were 350oC and 4.0 L/min. Full mass spectra were taken in the mass range of 50 to 1000, and the step size was 0.1 m/z. System control and data evaluation were conducted by using HP ChemStation.
1H/
Reaction optimization
To validate the commercial practicability of this approach, soybean lecithin was used for reaction optimization. Effects of traditional solvents, time, pH, concentration of PC and Vitamin B6, and enzyme efficiency were investigated, respectively. To facilitate comparison and evaluation, all performances were done under other identical or similar conditions. The conversion was indicated by the consumption of PC and the hydrolysis degree was expressed as the area percentage of PA in total phosphatidyl-based area from TLCFID data.
TLC-FID analysis
The samples from the reaction mixture was extracted by CHCl3- MeOH (3/1, v/v) and the resulting extract was washed with deionized water to remove pyridoxine. The TLC-FID analysis was performed on an Iatroscanner (Iatroscan MK6s, Iatron Laboratories, Tokyo, Japan) after the loaded samples were developed by solvent mixture of CHCl3-MeOH- H2O (50/20/3, v/v/v). PC-pyridoxine, PC and PA gave Rf of 0.71, 0.28 and 0.17, respectively. Area percentage was used as mass for the calculation of conversion of PC and yield of product.
Results and Discussion
Synthesis and characterization of DPPC-VB6
Preparation of DPPC-pyridoxine conjugates: To acquire a pure phosphatidyl derivative of pyridoxine for structural identification, DPPC has been used as a model PC to synthesize phosphatidyl VB6. The formation of a new product has been observed by the comparison of the TLC profiles of the reaction mixture before and after reaction (Figure 1). A new peak with Rf value at 0.71 excluding PC 0.28 and PA 0.17, as well as its time-dependent increase against the decrease of PC were observed. A typical time course monitored by TLC analysis was depicted in (Figure 2). As observed, the transphosphatidylation of DPPC to alcohol donor pyridoxol underwent a slow induction stage where the hydrolysis of DPPC into PA and transphosphatidylation of DPPC into PC-VB6 are almost equally in quantity. This probably can be ascribed the slow solublization of DPPC After 4h the transphosphatidylation increase linearly and becomes prevailing after 10h. The result seems suggested that 24 h is enough to maximize the reaction. Prolonging the reaction yields little beneficial effect on the preparation of DPP-VB6 rather more hydrolysis to PA.
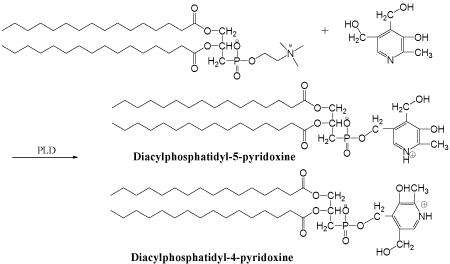
Figure 1: Schematic representation of PLD-catalyzed transphophatidylation
of pyridoxine.
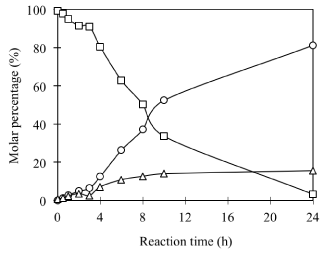
Figure 2: Time course of PLD catalyzed phosphatidylation of PC with VB6
(Symbols: square, DPPC; cycle, PC-VB6; triangle, PA). Other reaction
conditions: 30oC; 200μl PLD (2*9.3 U); 2mL ditheyl ether/2mL aqueous
phase; pH 5.6; Ca2+ 40 mM; pyridoxol 0.2*1.25/2mL= 125mM; DPPC=
0.05/2=25 mM.
Spectral analysis and structural conformation: As shown
in (Figure 1), phosphatidylation of pyridoxine could yield two
possible products: 4’-(1,2-dipaltitoylphosphatidyl) pyridoxine and
5’-(1,2-dipaltitoylphosphatidyl) pyridoxine (abb. DPP-VB6). The
API mass spectrum of the product showed the molecular ionic
peak [M-1]-, 798.5; [M-H2O-1]-1, 780.5; [M-pyridine (VB6)-1]-,
647.4; [M-palmityl-1]-, 542.2; [M-pyridine (VB6) palmityl-1]-, 391;
and [palmityl]-1, 255.2. The 1HNMR spectrum was as follows: 8.61
(1H, s, pyridium CH-6), 5.29 (2H, s, CH2-8), 4.67 (1H, s, aromatic
C-OH), 4.58 (2H, s, CH2-7), 4.24 (1H, m, glycerol CH-2), 4.16 (2H,
m, glycerol CH2-1), 4.06 (2H, m, glycerol CH2-3), 2.55 (3H, s, CH3),
2.25 (4H, m, palmitoyl CH2), 1.96 (1H, m, alcohol –OH), 1.58 (4H,
m, palmitoyl CH2), 1.25 (48H, m, CH2), 0.88 (6H, m, CH3). The
aforementioned mass spectrum and 1HNMR spectrum have identified
the conjunction of DPPC and VB6, but cannot identify the regioposition
of phosphatidyl linked.
![]()
Dipalmitoyl glycerol moiety
Pyridoxine moiety
CH3
13.06-13.43
CH2
18.71-33.84
CH2
35.83-36.40
C=O
178.63
CH2O
64.89
CHO
69.71
CH2OP
69.71
POC*H2-C
69.71
POCH2C
128.99
C-C*H-N
127.81
N-C*-CH3
131.26
CH3
15.92
C*-OH
166.74
C=C*-C
146.1
CH2OH
53.4
Table 1: Chemical shifts of 13CNMR of 1, 2-dipalmitoylphosphatidyl pyridoxine.
Optimization of reaction protocol
Effects of solvents: The paramount importance of solvents for PLD-catalyzed transphosphatidylation, which influences not only the total reaction rate but also the yield of desired product, has been intensively demonstrated [18,19]. Therefore, solvents having different hydrophobicity (log P) were screened. The significant effects of solvents on the phosphatidylation of pyridoxine in this work could be observed by the comparison of the reaction performance in different solvent/acetate buffer systems (Table 2). Based on the conversion of PC and hydrolysis degree, apart from water, the solvents used in this study could be classified into three groups. For the reaction without solvent addition, PC was dispersed by emulsion in buffer. Hydrolysis dominates the reaction, in which only 24% in total 80% conversion of PC is to form PC-VB6. Hydrophobic hexane and hydrophilic tert-butanol represent the first group, in which PLD show very low total activity, synthesis and hydrolysis activity. The reactions in the second group (in italic) showed bigger total activity; however, the considerable hydrolysis resulted in a low quality of the total reaction. The reactions in diethyl ether, chloroform and dichloromethane as the third group, showed preferable results, higher yield of PC and lower hydrolysis.
![]()
Yield of
PC
Hydrolysis
Log P
PC-VB6
conversion
degree
Water
24.2
79.1
54.9
-
tert-Butanol
7.6
10.5
2.9
0.35
n-Hexane
3.6
5.8
2.2
3.5
t-Butylmethyl ether
47.5
89.6
42.1
2.6
Ethyl acetate
54.8
97
42.2
0.68
Ethyl ether
71.2
96.9
25.7
0.85
Chloroform
84
97
13
2
Dichloromethane
83.7
97.7
14
1.25
*Other reaction conditions: temperature: 30oC; 200μl PLD (2*9.3 U); 2mL solvent/2mL aqueous phase; pH 5.6; Ca2+ 40 mM; pyridoxol 0.2*1.25/2mL= 125mM; soybean PC = 0.05/2=25 mM.
Table 2: Comparison of the reaction performances in different solvents*.
To interpret the effects of organic solvents on PLD catalyzed transformation in emulsion system, Ulbrich-Hofmann and coworkers [18] have suggested a convincible hypothesis that different effects of solvents result from the differing aggregate states of phospholipids in the interface of solvent/buffer where the reaction is assumed to take place. The authors established some correlation of interfacial pressure and activation of enzyme, as well as intercalation of some aliphatic alcohols to n-hexane, resulting in a significant increase of activity [17,18].
Generally, our results in preparation of PC-VB6 agree very well with the above hypothesis. However, the main goal is to set up an efficient system to acquire maximum desired product. Therefore minimizing hydrolysis is another important criterion to evaluate the system. According to our examination, a good and relative stable emulsion can always be observed among the second and third groups. Different from the reaction in the solvent of the second groups, the reaction in the solvent of third group could achieve a faster phase separation or form clear interface in shorter time. We suggested that this might be one of the reasons accounting for their less hydrolysis. It is well known that the PLD-catalyzed reaction takes place in the interface of organic solvent (dissolving phospholipids) and buffer (employed as a reservoir for water-soluble alcohols and the released choline). A good emulsion is undoubtedly necessary for the enzyme to have more efficient interaction with substrates. However, too long time for d-emulsification might mean prolonging hydrolysis time, because the concentration of alcohol (here is VB6) in the vicinity of emulsion and bulky phase maybe not evenly distributed. That is to say; too stable emulsion might hinder the transportation of alcohol to the vicinity of enzyme to compensate the consumed substrate, which results in the increase of local water activity. If the assumption is corrected, this effect should be substrate structuredependent but independent of enzyme source, because the physical properties of substrates and solvents govern the transportation. This is in coincidence with previous observation [18]. Of course the above hypothesis needs further experimental support.
Since the reaction in chloromethane gave the maximal yield of PC-VB6 and minimum hydrolysis degree, chloromethane was chosen as a model solvent in the following reaction optimization.
Comparison of DPPC and soybean PC for transphosphatidylation: The physical properties (melting point and solubility in organic solvent) of phosphatidylcholine are largely dependent on the fatty acid (chain length and unsaturated degree) composition. DPPC has a melting point of 41oC, while the soybean PC is semi-solid at room temperature, which can be easily dispersed and solubilized in most organic solvents as observed in this study. As observed in (Figure 3B), no obvious induction period is observed for soybean PC as for DPPC (Figure 3A). This difference can probably be attributed to better solubility of soybean PC in dichloromethane and their better dispersion in aqueous phase; which enable a better mixed emulsion and larger interface; thus promoting reaction. The time to reach equilibrium for soybean PC needs about 10h; whereas for DPPC it needs more than 24h. It is also evident that soybean PC achieves higher total conversion (>96%) than DPPC (60%); surprisingly, very little accumulation of PA for DPPC reaction was observed; indicating most PA further converts into diacylglycerol. The reason remains to be explored. Nevertheless, the ratios of main product/byproducts for DPPC and soybean PC are almost the same under other identical conditions (Figure 3A & 3B).
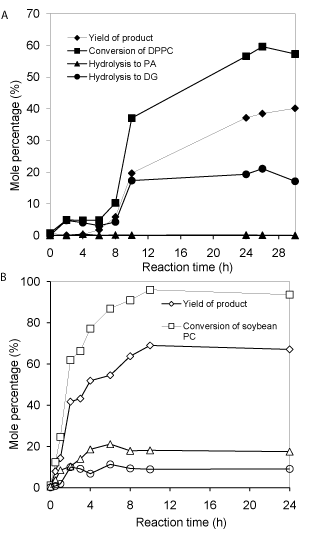
Figure 3: Comparison of PLD-catalyzed transphosphatidylation of DPPC (A)
and soybean PC (B). Other reaction conditions: 0.5 mmol DPPC or soybean
PC in 10 mL dichloromethane; 15*0.5 mmol VB6 in 10mL pH 5.6 buffer
containing 40 mM Ca2+; 30°C and PLD loading 40.4U.
Effects of pH: Examination of the effects of pH values has been performed in chloromethane/citric-diethylbarbituric buffer system with broad spectrum of pH (Figure 4). As shown in (Figure 4), PLD exhibited similar higher total activity (transphosphatidylation plus hydrolysis) at the range of pH 3.5-6.6. However, more hydrolyzed product yielded in more acidic environment, and quite low activity was observed at higher pH for both transphosphatidylation and hydrolysis. Better yield of desired product was expected to obtain at the range of pH 4.5-6.6, and the optimum pH is at around 5.4, since at which both maximal phosphatidylation activity and ratio of transphosphatidylation to hydrolysis rate was achieved. This result agreed well with the producer recommended value, but not the previous observations that PLD has a higher affinity for the nonionized form of substrates [17]. The non-ionized form of VB6 is a free base, a medium strong base, with lower solubility in aqueous solution, with which very low activity is attained. This result seems to suggest that the synthesis activity for transphosphatidylation was mainly governed by the nature of PLD.
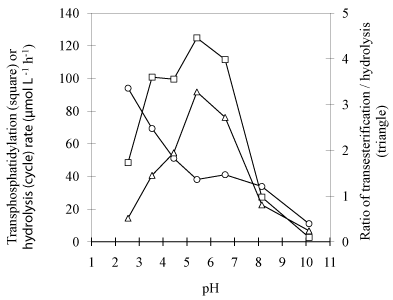
Figure 4: Effect of pH on PLD-catalyzed transphosphatidylation of DPPC to
VB6 and hydrolysis of DPPC. Other reaction conditions: Temperature 30°C,
reaction time 24h, 47mM DPPC and 0.7M VB6 and PLD loading 3.3 U/mL.
Effects of PC concentration: A proper substrate concentration is important to obtain higher yield at the desired time. Conversion of PC to PC-VB6 was therefore carried out at varying concentrations of PC using PLD in diethyl ether/buffer system (Figure 5). The results showed that at PC concentration of 15-25 mM over 90% conversion of PC (Figure 5A) and higher 80% yield of PC-VB6 (Figure 5B) could be achieved after 10h reaction with enzyme loading of 9.3U in 2 mL. So 3.0-5.0 mM PC/U PLD could be a recommended dosage for a scaled-up preparation of PC-VB6.
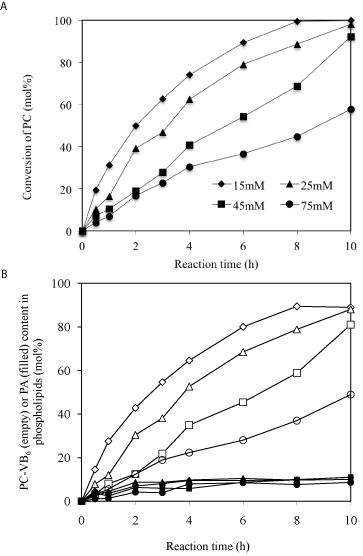
Figure 5: Effect of soybean PC concentration (cycle, square, triangle and
diamond stand for 15, 25, 45 and 75 mM, respectively). Other reaction
conditions: temperature: 30°C; 2mL diethyl ether; 2mL solvent/2mL aqueous
phase; pH 5.6; Ca2+ 40 mM; pyridoxol 0.3*1.25/2mL= 175.5mM; PLD; 100μL
(9.3U).
Effects of pyridoxine concentration: To examine the influence of excessive amount of pyridoxine on the yield of PC-VB6, the reactions with different pyridoxine concentrations have been conducted as shown in (Figure 6). Among the tested range, higher conversion of PC was obtained for all reactions. However, the hydrolysis of PC is relatively faster than transphosphatidylation, resulting in lower yield of PC-VB6. A significant increase of the yield of PC-VB6 against the enhancing concentration of pyridoxine clearly showed that excessive pyridoxine could suppress the hydrolysis of PC. In another word, higher VB6 concentration favors the synthesis of PC-VB6. The results in (Figure 6) displayed that higher ratio of pyridoxine/PC over 10 may be a better option to receive a higher yield of PC-VB6.
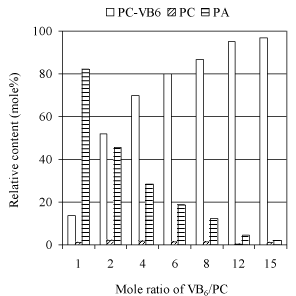
Figure 6: Effect of VB6 concentration. Other reaction conditions: temperature:
30°C; Reaction time 24h; 2mL diethyl ether; 2mL solvent/2mL aqueous
phase; pH 5.6; Ca2+ 40 mM; 47mM soybean PC; PLD 100uL (9.3U).
Effects of enzyme loading and enzyme efficiency: Figure 7A presented the effects of enzyme loading on conversion of PC and preference for synthesis. At the test range of enzyme concentrations, linear relationship between the reaction rate and enzyme load was observed in the first 10h. Similar specific activities (0.97-0.11 mmol/L h-1 U-1) were obtained, indicating no enzyme saturation is reached. However, the reactions with higher enzyme loading show higher selectivity for synthesis with prolonged time (Figure 7B). The results demonstrated that around 10 U/mL is enough for the reaction performed with 25 mM of PC in diethyl ether/buffer system to achieve preferable selectivity and enzyme efficiency.

Figure 7: Effect of enzyme loading. (A) Conversion of soybean PC and
yield of PC-VB6 with different PLD loading (triangle, cycle, square and
diamond, stand for the PLD concentration of 2.33, 4.65, 9.30 and 13.95 U/
mL; respectively); (B) Mole ratio of PC-VB6/PA at different reaction time
(Square (10h) and cycle (24h)). Other reaction conditions: temperature: 30°C;
In 2mL diethyl ether; 2mL solvent/2mL aqueous phase; pH 5.6; Ca2+ 40 mM;
pyridoxol 0.2*1.25/2mL= 125mM; PC= 0.05/2=25 mM.
Hydrolysis susceptibility of synthetic PC-VB6: We compared the hydrolysis activity of PC-VB6 to PA+VB6 and PC to PA (Figure 8) by PLD. It is obvious that PC undergoes a fast hydrolysis, which the hydrolysis can be completed in 2h; however, under the identical conditions the enzymatic hydrolysis of PC-VB6 precedes only 5% in 8h. This indicates a high resistance of phosphatidyl alcohol derivatives to enzymatic hydrolysis; which has been observed in other study [20]. The low hydrolysis susceptibility of synthetic PC-VB6 may lead to the slow release of VB6 in vivo, which can keep longer circulation time in human body, which might represent another interesting property of synthetic PC-VB6.
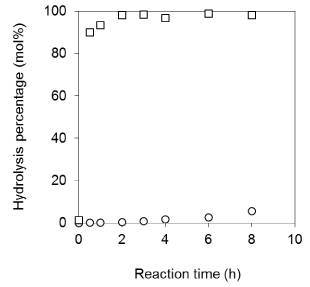
Figure 8: Comparison of the hydrolysis activities of soybean PC and PC-VB6
in ethyl acetate-buffer system. Other conditions: Organic phase 0.05 mmol
soybean PC and PC-VB6 in 2 mL ethyl acetate. Aqueous phase 2 mL of 100
mM acetate buffer, pH 5.6 containing 40 mM Ca2+ and 100 uL (10.1U).
Conclusion
In conclusion, this work reported a synthesis procedure of
a novel compound phosphatidyl pyridoxol (PC-VB6) by PLDcatalyzed
transphosphatidylation and the molecular structure of
the synthetic PC-VB6 were confirmed by MS, 1H/
References
- Stryer L. Biochemistry. 3th edn. New York: W. H. Freeman and Company. 1988.
- Bender DA. Vitamin B6. Nutr Food Sci. 1997; 4: 128-133.
- Compton MM, Cidlowski JA . Vitamin B6 and glucocorticoid action. Endocr Rev. 1986; 7: 140-148.
- Leklem JE, Brown RR, Rose DP, Linkswiler H, Arend RA. Metabolism of tryptophan and niacin in oral contraceptives users receiving controlled intakes of vitamin B6. Am J Clin Nutr. 1975; 28: 146-156.
- Zhao M, Lamers Y, Ralat MA, Coats BS, Chi YY, Muller KE, et al. Marginal vitamin B-6 deficiency decreases plasma (n-3) and (n-6) PUFA concentrations in healthy men and women. J Nutr. 2012; 142: 1791-1797.
- Molina A, Oka T, Munoz SM, Chikamori-Aoyama M, Kuwahata M, Natori Y. Vitamin B6 suppresses growth and expression of albumin gene in a human hepatoma cell line HepG2. Nutr Cancer. 1997; 28: 206-211.
- Komatsu S, Yanaka N, Matsubara K, Kato N. Antitumor effect of vitamin B6 and its mechanisms. Biochim Biophys Acta. 2003; 1647: 127-130.
- Mindell E. The vitamin bible: how the right vitamins and nutrient supplements can revolutionise your life. 2nd edn. London: Arlington Books, 1998.
- Bruttomesso AC, Tiscornia A, Baldessari A. Lipase-catalyzed preparation of biologically active esters of dehydroepiandrosterone. Biocat Biotrans. 2004; 22: 215-220.
- Guo Z, Vikbjerg AF, Xu X. Enzymatic modification of phospholipids for functional applications and human nutrition. Biotechnol Adv. 2005; 23: 203-259.
- Jo DH, Abdallah MA, Young J, Baulieu EE, Robel P. Pregnenolone, dehydroepiandrosterone, and their sulfate and fatty acid esters in the rat brain. Steroids. 1989; 54: 287-297.
- Baldessari A, Mangone CP, Gros EG. Lipase-Catalyzed Acylation and Deacylation Reactions of Pyridoxine, a Member of Vitamin-B6 Group. Helv Chim Acta. 1998; 81: 2407-2413.
- Fricker G, Kromp T, Wendel A, Blume A, Zirkel J, Rebmann H, et al. Phospholipids and lipid-based formulations in oral drug delivery. Pharm Res. 2010; 27: 1469-1486.
- Li J, Wang X, Zhang T, Wang C, Huang Z, Luo X, et al. A review on phospholipids and their main applications in drug delivery systems. 2015; 10: 81–98.
- Servi S. Phospholipases as synthetic catalysts. Top Curr Chem. 1999; 200: 127–158.
- D'Arrigo P, Servi S. Using phospholipases for phospholipid modification. Trends Biotechnol. 1997; 15: 90-96.
- Ulbrich-Hofmann R. Enzyme-catalyzed transphosphatidylation. Eur J Lipid Sci Technol. 2003; 105: 305–308.
- Hirche F, Ulbrich-Hofmann R. The Interdependence of Solvent. Acceptor Alcohol and Enzyme Source in Transphosphatidylation by Phospholipase D. Biocatal Biotransformation. 2000; 18: 343-353.
- Wang P, Schuster M, Wang YF, Wong CH. Synthesis of phospholipid inhibitor conjugates by enzymatic transphosphatidylation with phospholipase-D. J Am Chem Soc.1993; 115: 10487–10491.
- Nagao A1, Ishida N, Terao J. Synthesis of 6-phosphatidyl-L-ascorbic acid by phospholipase D Lipids. 1991; 26: 390-394.
- Takami M, Hidaka N, Miki S, Suzuki Y. Enzymatic-synthesis of novel phosphatidylkojic acid and phosphatidylarbutin, and their inhibitory effects on tyrosinase activity. Biosci Biotechnol Biochem. 1994; 58: 1716– 1717.