
Letter to the editor
Austin J Gastroenterol. 2014;1(2): 1008.
Persistent Vomiting in an Adolescent – Not a Crying Matter
Aaron Johnston1 and Michael J Nowicki2*
1Department of Pediatrics, University of Mississippi Health Center, USA
2Department of Pediatric Gastroenterology, University of Mississippi Health Center, USA
*Corresponding author: :Michael J Nowicki, Department of Pediatric Gastroenterology, University of Mississippi Health Center, 2500, North State Street, Jackson, MS, 39216, USA
Received: June 12, 2014; Accepted: June 25, 2014; Published: June 27, 2014
We report an adolescent with Triple-A syndrome (Addison�s disease, achalasia, and alacrima) to remind your readers of this rare condition. A 13-year-old male presented with a six month history of non-bilious, non-bloody post-prandial vomiting. He denied associated abdominal pain, headaches, and nocturnal vomiting. Past history was significant for Addison�s disease, prior hypoglycemic seizures, and scoliosis; his only surgery was repair of a tethered cord. Examination revealed a well-nourished male with marked scoliosis. Heart and lung examinations were normal. His abdomen was soft, non-tender, and without hepatosplenomegaly. Laboratory evaluation included normal values for complete blood count, erythrocyte sedimentation rate, liver panel, lipase, serum electrolytes, and glucose. A presumptive diagnosis of gastro esophageal reflux was made and he was started on a daily proton pump inhibitor.
At 6 month follow-up he had gained 1.9 kg despite persistence of vomiting. His examination remained unrevealing. An upper gastrointestinal series was obtained, showing findings suggestive for a stricture at the gastro esophageal junction causing almost complete obstruction (Figure 1). He was scheduled for esophagogastroduod endoscopy with balloon dilation for presumed peptic stricture the following day.
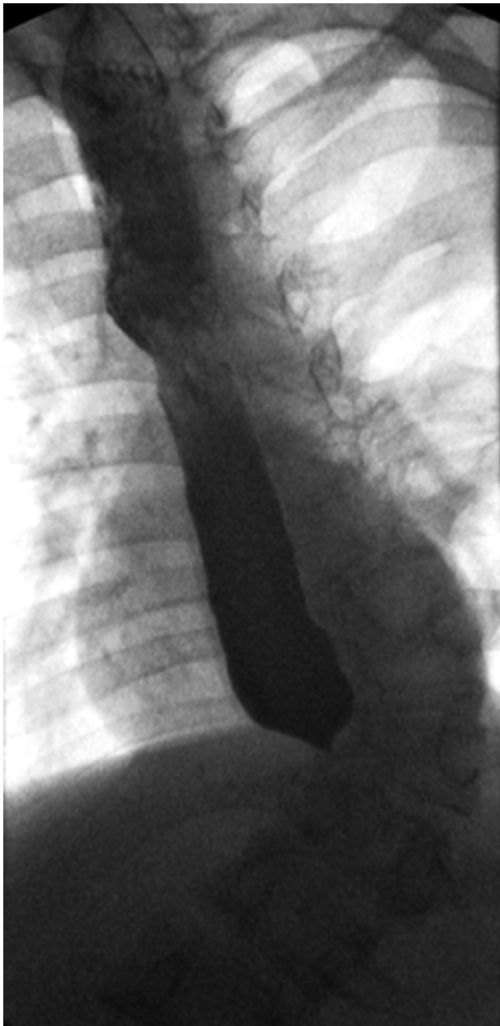
Figure 1: Barium esophagram showed a marked “bird-beaked” narrowing at the lower esophageal sphincter and dilation of the esophagus.
At endoscopy the esophagus was dilated with normal-appearing mucosa with retained specks of barium. The endoscope easily passed through the lower esophageal sphincter consistent with a diagnosis of achalasia. Dilation was performed using through-the-scope radial balloon dilators. Further history revealed that the patient had never cried tears. A diagnosis of Triple-A syndrome was rendered.
Heller myotomy was performed 2 months later, with complete resolution of symptoms. The patient has remained asymptomatic during a 24 month follow-up period.
Achalasia is a rare disorder with a prevalence of 0.5 to 1 per 100,000 per year with less than 5% of patients presenting prior to 15 years of age [1]. Achalasia arises due to denervation of inhibitory neurons within the enteric plexus leading to impaired relaxation of the lower esophageal sphincter and loss of esophageal peristalsis. Most cases of achalasia are considered idiopathic, although evidence is gathering to support an auto-immune response targeted to neurons [2]. Achalasia may occur in association with adrenal insufficiency and alacrima; an autosomal recessive condition referred to as Triple A syndrome or All grove syndrome. Less commonly described features of Triple A syndrome include autonomic nerve dysfunction, neuro degeneration, hyperkeratosis, osteoporosis, and microcephaly [3]. The gene responsible for Triple A syndrome (AAAS gene) is located on chromosome 12q13 [4]; the gene product is a 546 amino acid protein called alacrima-achalasia-adrenal insufficiency neurologic disorder (ALADIN) protein [5].
References
- Earlam RJ, Ellis FH Jr, Nobrega FT. Achalasia of the esophagus in a small urban community. Mayo Clin Proc. 1969; 44: 478-483.
- De Giorgio R, Guerrini S, Barbara G, Stanghellini V, De Ponti F, Corinaldesi R, et al. Inflammatory neuropathies of the enteric nervous system. Gastroenterology. 2004; 126: 1872-1883.
- Houlden H, Smith S, De Carvalho M, Blake J, Mathias C, Wood NW, et al. Clinical and genetic characterization of families with triple A (Allgrove) syndrome. Brain. 2002; 125: 2681-2690.
- Weber A, Wienker TF, Jung M, Easton D, Dean HJ, Heinrichs C, et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster. Hum Mol Genet. 1996; 5: 2061-2066.
- Handschug K, Sperling S, Yoon SJ, Hennig S, Clark AJ, Huebner A. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet. 2001; 10: 283-290.