
Special Article-Inflammatory Bowel Disease
Austin J Gastroenterol. 2014;1(4): 1020.
Analysis of Mucosal Microbiota in Inflammatory Bowel Disease using a Custom Phylogenetic Microarray
Arun Gupta1,2*, Seungha Kang3, Josef Wagner4, Carl Kirkwood4, Mark Morrison3, Chris McSweeney3 and Finlay Macrae5
1Gastroenterology and Hepatology Unit, the Canberra Hospital, Australia
2The University of Melbourne, Australia
3CSIRO Livestock Industries, St Lucia
4Enteric Viruses Unit, Murdoch Childrens Research Institute, Australia
5Department of Colorectal Medicine and Genetics, Royal Melbourne Hospital, Australia
*Corresponding author: : Arun Gupta, Department of Gastroenterology, The Canberra Hospital, Post Box: 11, Woden, Australian Capital Territory, 2606, Australia
Received: August 11, 2014; Accepted: September 09, 2014; Published: September 12, 2014
Abstract
Background and Aims: The pathogenesis of inflammatory bowel disease is likely to involve interaction between genetic factors, innate immunity, and the enteric microbiota. Alterations in the composition of the normal commensal microbiota may play a pathogenic role.
Methods: A custom 2240 probe oligonucleotide microarray based on 16s RNA sequences was used to compare the microbiota profiles of patients with inflammatory bowel disease with controls. Twenty mucosal samples obtained from colonoscopic biopsies were analysed - five from Crohn’s Disease Inflamed (CDI) tissue, five from Crohn’s Disease Non-Inflamed (CDNI), five from Ulcerative Colitis (UC), and five healthy control samples. Analysis was performed using principal components analysis and between group analysis.
Results: The microbiota from both Crohn’s disease and ulcerative colitis differed significantly from the control group, though not between CDI and CDNI groups. Alterations in the abundance of Faecalibacterium prausnitzii, Shigella flexneri, Dorea longicatena, and Xenorhabdus bovienii were associated with Crohn’s disease. Alterations in the abundance of Yersinia pestis and Eubacterium rectale were associated with ulcerative colitis.
Conclusion: The gastrointestinal microbiota differed in mucosal samples from patients with inflammatory bowel disease compared to those taken from controls. The composition of the microbiota was not altered by the presence of inflammation. The abundance of particular organisms including the previously described F. prausnitzii was found to be different in patients with inflammatory bowel disease compared to healthy controls, and new putative aetiological organisms were identified. These findings support the hypothesis that a bacterial ‘dysbiosis’ may contribute to the pathogenesis of inflammatory bowel disease.
Keywords: Microbiota; Microarray; Inflammatory bowel disease; Crohn’s disease; Ulcerative colitis
Introduction
Crohn’s disease and ulcerative colitis are inflammatory disorders of the gastrointestinal tract, which can cause significant morbidity in patients. Treatment is often with medications affecting the immune system, with the aim of reducing gastrointestinal inflammation. Advances in genetics and microbiology have shed light on factors that are likely to be playing a role in pathogenesis of these inflammatory bowel diseases. Single nucleotide polymorphisms affecting genes relating to the innate immune system have been associated with inflammatory bowel disease (IBD), including mutations of the NOD2, TLR4, IL23R and ATG16L1genes [1-4]. Functional studies have shown that some of these genes affect bacterial sensing within the gastrointestinal tract, possibly affecting immunological tolerance and disrupting the delicate homeostasis between the mucosal lining of the gut and the bacterial microbiota [5-8].
The role of the bacterial microbiota within the gastrointestinal tract in IBD is less well characterised. Many studies have suggested a role for individual organisms playing a role in initiation or perpetuation of IBD, such as Mycobacterium avium subspp. Paratuberculosis (MAP) and adherent-invasive Escherichiacoli (AIEC) [9-11]. Although found in increased abundance in patients with IBD in many studies, the evidence to support a pathogenic role for these organisms has been inconsistent, possibly due to differing tissues being analysed, differing specimen handling and analysis techniques, and different populations being studied. A multi-centre trial assessing patients with Crohn’s disease treated empirically for MAP with clarithromycin, rifabutin and clofazimine did not reach the primary outcome of decreased rate of relapse [12].
An alternative hypothesis to the single pathogen hypothesis is that alterations in the composition of the normal gastrointestinal microbiota (also termed ‘dysbiosis’) may play a pathogenic role in IBD. Traditional microbiological techniques have been based on culture of organisms on nutrient rich plates, however approximately 80% of organisms in the gastrointestinal tract are not able to be cultured ex vivo [13]. Molecular techniques based on the bacterial 16s ribosomal RNA ‘fingerprint’ can identify these culture negative bacteria, and can be used to categorise previously undiscovered bacteria into the bacterial taxonomy. Techniques to analyse the microbiota including Temporal Temperature Gradient gel Electrophoresis (TTGE), Denaturing Gradient Gel Electrophoresis (DGGE), Automated Ribosomal Intergenic Spacer Analysis (ARISA) to generate microbial community ‘profiles’, as well as metagenomic approaches where the entire library of organisms in a particular community are catalogued and described [13-17]. Different studies comparing the microbiota in IBD compared to controls have assessed the microbiota in mucosal samples, faecal samples and surgical resection specimens. The current study aims to assess the difference in microbiota in mucosal samples from patients with IBD compared to controls using a custom built 2240 probe microarray based on 40-mer 16s ribosomal RNA oligonucleotides.
Materials and Methods
Recruitment & Sample collection
Approval was obtained from the human research ethics committees at Melbourne Health, Melbourne Private Hospital and Cabrini Health (HREC.2006.267). Patients aged between eighteen and eighty years of age who were already scheduled for a colonoscopy were recruited after informed consent was obtained. Patients were recruited from two separate groups, the first comprising patients without a diagnosis of IBD who were planned for a colonoscopy to investigate diarrhoea, and the second group with a pre-existing diagnosis of IBD. Patient epidemiology was collected, and phenotype was recorded if the patient was known to have IBD.
Mucosal biopsies were collected at the time of colonoscopy from both inflamed and non-inflamed areas. Biopsy specimens were transferred immediately to tubes containing RNA later buffer (Applied Bio systems, Austin, Texas, USA) and then frozen. DNA extraction was later performed from these samples in accordance with previously described methods [18]. A total of 109 samples were collected from 35 patients. A subset of samples were analysed as part of this study; five samples from the Crohn’s Disease Inflamed (CDI) group, five from Crohn’s Disease Non-Inflamed (CDNI), five from Ulcerative Colitis (UC) and five from the control group. The control group comprised of patients who had a normal colonoscopy and normal histopathology.
Oligonucleotide probe design and microarray preparation
Between group analyses between all four groups
A custom phylogenetic microarray was designed and validated according to the methods described by Kang et al. (2010) [19]. Briefly, the probes based on 16s ribosomal RNA sequences from gastrointestinal bacteria identified on the Entre z Nucleotide database situated on the National Centre for Biotechnology Information (NCBI) website were included. The particular oligonucleotides selected were derived from a literature search of published papers [20,21] Individual probes were then designed using the Go Array oligonucleotide program [22]. The resultant 40-mer oligonucleotide probes represented diverse taxonomy groups, with differing specificities ranging from species to phylum levels; the majority were targeted at the species level. These were synthesised on to a custom 4X2K probe microarray by Combimatrix (USA). The microarray was synthesized with four replicates of each probe distributed across the array to allow validation of results. Samples were analysed as per the Combimatrix hybridisation and imaging protocol. Resultant microarray images were analysed using Gene Pix Pro 6.0 software [23,24].
Statistical analysis
The microarray data were normalized using the quantile normalization method, then analysed using the R statistical environment [25,26]. The four different groups were compared using betweengroup analysis (BGA), correspondence analysis (CoA), and Monte-Carlo tests. These were performed using the multivariable analysis, ADE4 and MADE4 software packages [26-29].The analysis is classified as a ‘supervised’ microarray analysis in that different groups were defined beforehand. The Monte-Carlo permutation test was used to generate P values for differences between samples.
Results
20 samples were analysed using the microarray platform; 5 from the CDI group, five from the CDNI group, 5 from the UC group and 5 control samples.
Between group analyses between all four groups
Between Group Analysis (BGA) suggested significant separation between the four sample groups analysed, Crohn’s Disease Inflamed (CDI), Crohn’s Disease Non-Inflamed (CDNI), Ulcerative Colitis (UC) and controls when comparing all bacterial species (Figure 1). The principal components analysis of the four groups was analysed using a Monte Carlo test, giving a p-value of 0.005.
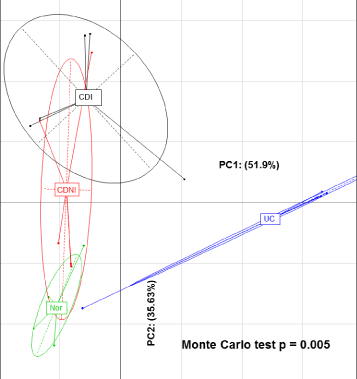
Figure 1: Between group analysis between Crohn’s Disease Inflamed (CDI), Crohn’s Disease Non-Inflamed (CDNI), Ulcerative Colitis (UC), and the control group (‘Nor’).
The abundance of each of the 2240 organism specific probes present in the microarray within the four different groups was compared using BGA, and employing using the Monte Carlo test. Of these comparisons, seven revealed statistical significance (p-value less than 0.05); Faecalibacterium prausnitzii (p = 0.046), Clostridum proteolyticum (p = 0.002), Clostridium butyricum (p = 0.001), Bacteroides thetaiotamicron (p = 0.018), Bacteroides distasonis (p = 0.003), Enterobacter cowanii (p = 0.027)and Proteus vulgaris (p = 0.013).Of note, both the Faecalibacterium and Clostridium species fall within the Clostridiaceae family.
Faecalibacterium prausnitzii was analysed specifically and was noted to be decreased in the CDI and CDNI subgroups compared to controls, and was decreased in the UC compared to all three (Figure 2). In the Clostridum proteolyticum species, the abundance in ulcerative colitis samples was greatly increased compared to samples from Crohn’s disease, CDI and CDNI (data not shown).
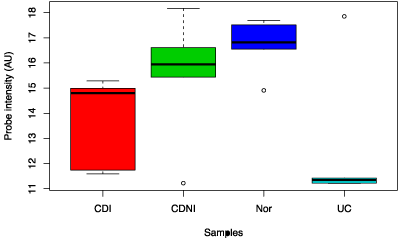
Figure 2: Box chart analysis comparing abundance of Faecalibacterium prausnitzii in different groups (P=0.04).
Comparison between Crohn’s disease (inflamed) and control group
The Crohn’s disease (inflamed) and the control groups were compared using a heat map of the top ten probes showing the strongest association, based on the BGA. The BGA suggested a difference between the two groups overall (p = 0.033), and the abundances of Faecalibacterium prausnitzii, Bacteroides vulgatus, Dorea longicatena, Bacteroides caecae and All stipes putredines appeared different in abundance between the two groups on the heat map (Figure 3). However, when the relative abundances of organisms were assessed individually, only the abundances of Faecalibacterium prausnitzii and Dorea longicatena were noted to be decreased in Crohn’s disease compared to controls, and Xenorhabdus bovienii and Shigella flexneri were increased in abundance in the Crohn’s group compared to control samples (see Figure 4 and Figure 5).
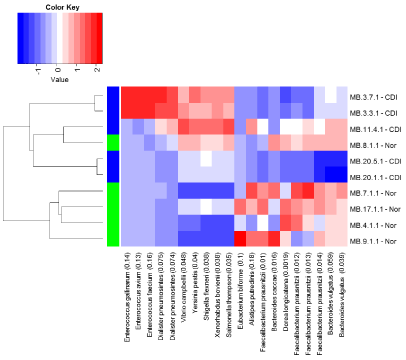
Figure 3: Heat map analysis comparing CDI and control groups.
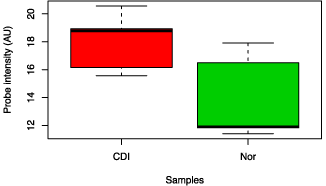
Figure 4: Box chart analysis comparing abundance of Shigella flexneri between CDI and control groups (p value = 0.04).
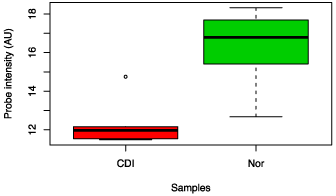
Figure 5: Box chart analysis comparing abundance of F. prausnitzii between CDI and control groups (p value = 0.02).
Comparison between samples from ulcerative colitis and control groups
In the heat map analysis of the top ten probes based on the BGA comparing samples from patients with ulcerative colitis compared to control patients, differences in abundances were seen in a number of organisms including Bacteroides thetaiotamicron, Faecalibacterium prausnitzii, Bacteroides distasonis, Dorea longicatena and Bacteroides fragilis (see Figure 6). BGA suggested that the difference between the groups was statistically significant with a p-value of 0.039. When organisms were compared individually, the abundance of only two organisms was noted to be statistically significant; Yersinia pestis and Eubacterium rectale (see Figure 7 and Figure 8). The difference in the abundance of F. prausnitzii between UC and controls on the heat map was p = 0.019 using one probe, and p = 0.14 using another probe, and did not reach statistical significance when individually compared. This is despite the striking difference in the initial four group comparison (see Figure 2).
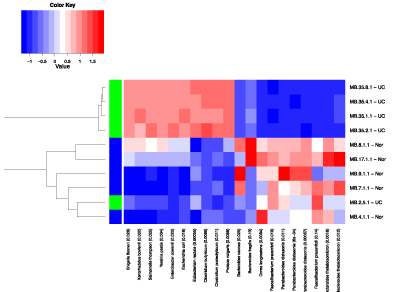
Figure 6: Heat map analysis comparing Normal and Ulcerative colitis (UC) samples.
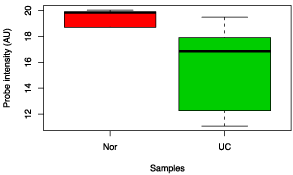
Figure 7: Box chart analysis comparing abundance of Eubacterium rectale between UC and control groups (p value = 0.04).
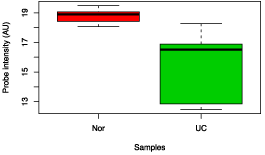
Figure 8: Box chart analysis comparing abundance of Yersinia pestis between UC and control groups (p value = 0.02).
Comparison between Crohn’s disease inflamed and Crohn’s disease non-inflamed samples
A heat map based group analysis between Crohn’s disease inflamed samples and Crohn’s disease non-inflamed samples suggested differences in abundance of particular organisms, however, this analysis did not detect a statistically significant different between the two groups as a whole (p = 0.30). As such, no subgroup analysis was performed between these two groups of samples.
Discussion
This study shows significant differences in the bacterial profiles of the mucosal microbiota between patients with either CD or UC and controls, based on analysis of mucosal samples using a custom 16s rRNA oligonucleotide probe microarray. The use of the oligonucleotide microarray allowed analysis of specimens across a large range of species, and also provided a semi-quantitative analysis of abundance to be performed. Comparing all four groups (CDI, CDNI, UC and controls) using principal components analysis, suggested that a small number of the differences identified between the groups were statistically significant. This analysis showed significant differences between the groups in a number of organisms including members of the Bacteroides, Faecalibacterium, and Clostridium subgroups.
Subsets of two groups were then compared directly using a heat map based on the BGA. Samples from CDI were different to control patients, reaching statistical significance for several organisms. The abundance of F.prausnitzii and Dorea longicatena was decreased in the CDI group compared to controls, and the abundance of Xenorhabdus bovienii and Shigella flexneri was increased in the CDI group compared to controls. Shigella flexneri is a Gram negative organism, of the family Enterobacteriaceae which is known as a gastrointestinal pathogen in humans causing a diarrhoeal illness. (30) Xenorhabdus (also known as Photorhabdus) are Gram negative gamma proteobacteria which are better known as colonising the guts of soil nematodes [31].
In the current study, samples from patients with ulcerative colitis were compared to those from control patients, and the BGA suggested statistically significant differences between the two groups. When the organisms were compared specifically, the abundances of Yersinia pestis and Eubacterium rectale were noted to be decreased in the UC group compared to controls. The difference in the abundance of F. prausnitzii was not statistically significant between the two groups overall. When the CDI and CDNI groups were compared using heat map based on the between group analysis, a number of organisms including F. prausnitzii appeared different in abundance, however the BGA did not suggest a statistically significant difference between these two groups.
The major findings of this study is that the microbiota in Crohn’s disease and ulcerative colitis groups appeared to be different from the control group, and that no difference was detected in Crohn’s disease samples between inflamed samples and non-inflamed samples. This argues against the hypothesis that inflammation is the primary cause of alterations in the microbiota. A primary ‘field’ effect in IBD may contribute to pathogenesis, however the possibility that previous inflammation or inflammation in other parts of the bowel may have contributed to the dysbiosis still needs to be considered. Another major finding was that the identification of organisms not previously associated with IBD, which displayed differential abundance between IBD and control patients. Shigella flexneri, Dorea longicatena, and Xenorhabdus bovienii were associated with Crohn’s disease, and Yersinia pestis and Eubacterium rectale were associated with ulcerative colitis.
This study confirms previously noted findings that F. prausnitzi I is decreased in abundance in patients with Crohn’s disease compared to controls. This association was not seen in patients with UC compared to controls. Sokol et al (2008) noted that patients with ileal Crohn’s disease who underwent surgical resection were more likely to have post-operative recurrence if the abundance of F. prausnitzii was decreased [32]. Fujimoto et al. [33] found decreased F. prausnitzii in the stool samples of patients with Crohn’s disease compared to controls, using terminal restriction fragment length polymorphisms and real time polymerase chain reaction [33] F. prausnitzii is a known butyrate producer, and butyrate may have ‘anti-inflammatory’ properties, possibly due to decreased proinflammatory cytokine expression via inhibition of nuclear factor kappa B activation and degradation of the I kappa B alpha protein [34,35].
The findings from this study do support the hypothesis that alterations in the enteric microbiota contribute to the pathogenesis of IBD. The individual organisms identified may form part of this ‘dysbiosis’, or alternatively they may represent individual ‘pathogens’. This raises the question of whether a normal commensal or ‘allochthonous’ organism may become ‘pathogenic’ if their abundance is increased or decreased significantly compared to normal variation [36]. Although the sample sizes were small in this study, the redundancy in the probes in the microarray and the magnitude of the differences in abundance make the results more robust. Nonetheless, these findings need to be validated in larger sample sets. Another potential limitation is that control patients may have had a diagnosis of irritable bowel syndrome as they had a normal colonoscopy to investigate diarrhoea. As such their microbiota may be different compared to healthy volunteers.
Whether the alterations in the relative abundance of particular organisms or whether a broader dysbiosis is thought to be more important, therapeutic options which directly affect the microbiota include antibiotics, prebiotics, probiotics or even faecal transplantation. ‘Prebiotics’ refer to dietary oligosaccharides that may alter the abundance of particular organisms. Together with probiotics these are termed together as ‘synbiotics’[37]. Apart from a few specific situations such as probiotics following pouch surgery for colitis or antibiotics for perianal Crohn’s disease, the evidence to support the use of these therapeutic modalities in inflammatory bowel disease is lacking [38,39]. At this point in time, the majority of treatments used in IBD reduce inflammation by their effect on the immune system. Whether these treatments also affect the bacterial microbiota directly remains an interesting possibility.
Acknowledgement
AG and FM conceived the study, contributed to study design and coordination and collected colonoscopic biopsies. JW and CK carried out DNA extraction on the samples and contributed to study design. SK, CM, and MM designed the custom oligonucleotide array and carried out the sample analysis. All authors read and approved the final manuscript. This project was supported by the Victorian Operational Infrastructure Support Program.
References
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001; 411: 599-603.
- Franchimont D, Vermeire S, E Housni H, Pierik M, Van Steen K, Gustot T, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004; 53: 987-992.
- Cummings JR, Cooney R, Pathan S, Anderson CA, Barrett JC, Beckly J, et al. Confirmation of the role of ATG16L1 as a Crohn's disease susceptibility gene. Inflamm Bowel Dis. 2007; 13: 941-946.
- Brand S, Staudinger T, Schnitzler F, Pfennig S, Hofbauer K, Dambacher J, et al. The role of Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms and CARD15/NOD2 mutations in the susceptibility and phenotype of Crohn's disease. Inflamm Bowel Dis. 2005; 11: 645-652.
- van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn's disease. Lancet. 2005; 365: 1794-1796.
- Tsay TB, Chang CJ, Chen PH, Hsu CM, Chen LW. Nod2 mutation enhances NF-kappa B activity and bacterial killing activity of macrophages. Inflammation. 2009; 32: 372-378.
- Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013; 62: 1505-1510.
- Arulampalam V, Greicius G, Pettersson S. The long and winding road to gut homeostasis. Curr Opin Gastroenterol. 2006; 22: 349-353.
- Autschbach F, Eisold S, Hinz U, Zinser S, Linnebacher M, Giese T, et al. High prevalence of Mycobacterium avium subspecies paratuberculosis IS900 DNA in gut tissues from individuals with Crohn's disease. Gut. 2005; 54: 944-949.
- Kuenstner JT. Mycobacterium avium subspecies paratuberculosis: a human pathogen causing most cases of Crohn's disease. Am J Gastroenterol. 2006; 101: 1157-1158.
- Rolhion N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2007; 13: 1277-1283.
- Selby W, Pavli P, Crotty B, Florin T, Radford-Smith G, Gibson P, et al. Antibiotics in Crohn's Disease Study G. Two-year combination antibiotic therapy with clarithromycin, rifabutin, and clofazimine for Crohn's disease. Gastroenterology. 2007; 132: 2313-2319.
- Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008; 3: 417-427.
- Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC microbiology. 2011; 11: 7.
- Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009; 15: 1183-1189.
- Sepehri S, Kotlowski R, Bernstein CN, Krause DO. Microbial diversity of inflamed and noninflamed gut biopsy tissues in inflammatory bowel disease. Inflamm Bowel Dis. 2007; 13: 675-683.
- Lepage P, Leclerc MC, Joossens M, Mondot S, Blottière HM, Raes J, et al. A metagenomic insight into our gut's microbiome. Gut. 2013; 62: 146-158.
- Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004; 36: 808-812.
- Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, et al. Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010; 16: 2034-2042.
- Wang RF, Beggs ML, Robertson LH, Cerniglia CE. Design and evaluation of oligonucleotide-microarray method for the detection of human intestinal bacteria in fecal samples. FEMS Microbiol Lett. 2002; 213: 175-182.
- Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, et al. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Research. 2006; 34: e42.
- Rimour S, Hill D, Militon C, Peyret P. Go Arrays: highly dynamic and efficient microarray probe design. Bioinformatics. 2005; 21: 1094-1103.
- Gene Pix Pro 6.0. Molecular Devices.
- Gene Spring, 7.3 ed. Agilent Technologies, Santa Clara, California.
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003; 19: 185-193.
- Culhane AC, Perrière G, Considine EC, Cotter TG, Higgins DG. Between-group analysis of microarray data. Bioinformatics. 2002; 18: 1600-1608.
- Fellenberg K, Hauser NC, Brors B, Neutzner A, Hoheisel JD, Vingron M. Correspondence analysis applied to microarray data. Proc Natl Acad Sci USA. 2001; 98: 10781-10786.
- Baty F, Facompré M, Wiegand J, Schwager J, Brutsche MH. Analysis with respect to instrumental variables for the exploration of microarray data structures. BMC Bioinformatics. 2006; 7: 422.
- Culhane AC, Thioulouse J, Perrière G, Higgins DG. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics. 2005; 21: 2789-2790.
- Philpott DJ, Edgeworth JD, Sansonetti PJ. The pathogenesis of Shigella flexneri infection: lessons from in vitro and in vivo studies. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2000; 355: 575-586.
- Forst S, Dowds B, Boemare N, Stackebrandt E. Xenorhabdus and Photorhabdus spp: bugs that kill bugs. Annu Rev Microbiol. 1997; 51: 47-72.
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008; 105: 16731-16736.
- Fujimoto T, Imaeda H, Takahashi K, Kasumi E, Bamba S, Fujiyama Y, et al. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn's disease. J Gastroenterol Hepatol. 2013; 28: 613-619.
- Hold GL, Schwiertz A, Aminov RI, Blaut M, Flint HJ. Oligonucleotide probes that detect quantitatively significant groups of butyrate-producing bacteria in human feces. Appl Environ Microbiol. 2003; 69: 4320-4324.
- Segain JP, Raingeard de la Blétière D, Bourreille A, Leray V, Gervois N, Rosales C, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn's disease. Gut. 2000; 47: 397-403.
- Tannock GW. What immunologists should know about bacterial communities of the human bowel. Semin Immunol. 2007; 19: 94-105.
- Hedin C, Whelan K, Lindsay JO. Evidence for the use of probiotics and prebiotics in inflammatory bowel disease: a review of clinical trials. Proc Nutr Soc. 2007; 66: 307-315.
- Gionchetti P, Rizzello F, Venturi A, Brigidi P, Matteuzzi D, Bazzocchi G, et al. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000; 119: 305-309.
- Komanduri S, Gillevet PM, Sikaroodi M, Mutlu E, Keshavarzian A. Dysbiosis in pouchitis: evidence of unique microfloral patterns in pouch inflammation. Clin Gastroenterol Hepatol. 2007; 5: 352-360.