
Mini Review
Austin J Gastroenterol. 2017; 4(1): 1076.
BRCA1 and BRCA2 Mutation in Pancreatic Cancer: Significance in Therapeutic Approach
Biswas AS¹ and Chakraborty A²*
¹Maulana Abul Kalam Azad University of Technology, India
²Division of Cellular & Molecular Biology, The Hormel Institute, USA
*Corresponding author: Chakraborty A, Cellular & Molecular Biology Division, The Hormel Institute- University of Minnesota, 801 16th Avenue NE, Austin, MN 55912, USA
Received: January 01, 2017; Accepted: February 27, 2017; Published: March 01, 2017
Abstract
Germline mutations in the tumour suppressor genes breast cancer antigen gene BRCA1 and BRCA2 have been proven to portend a drastically increased lifetime risk of breast and ovarian cancers in the individuals who carry them. A number of studies have shown that the third most common cancer associated with these mutations is pancreatic cancer. Pancreatic ductal adenocarcinoma (PDAC) remains one of the greatest challenges in oncology. Though it is estimated that about 5 percent of patients with pancreatic cancer are BRCA carriers, this subset of individuals may be more responsive to therapies that damage DNA, such as some chemotherapies, radiation therapy and some targeted therapies. As a result, BRCA carriers with pancreatic cancer may live longer than their counterparts who do not carry the mutation. We study the therapeutic approach and importance of BRCA1/2 mutation in pancreatic cancer.
Keywords: BRCA1; BRCA2; Pancreatic cancer; PARP; Olyperab
Introduction
Mutations in BRCA1 and BRCA2, most commonly linked with breast and ovarian cancers. But nowadays it was established that this genes are also associated with pancreatic cancer as well. A person with BRCA1 or BRCA2 mutations has a 5 percent risk to develop pancreatic ductal adenocarcinoma (PDAC) in their lifetime. Mutations in germ line level involving many genes that can lead to develop pancreatic cancer. Such genes are BRCA1, BRCA2 [1,2], TP53 [3], PALB2 [4], P16/CDKN2A [5,6], SMAD4 [7], STK11 [8], ataxia-telangiectasia-mutated (ATM) gene [9] and the mismatch repair genes (MMR) [10]. 5-10% are familial cancer [11-13]. BRCA1 and 2 are autosomal dominant genes having incomplete penitrance [14]. The tumor suppressor genes are those genes which controls cell growth and differentiation and drives tumorigenesis in a cascade pathway manner [15]. BRCA protein involves in post transcriptional protein expression as well a DNA double strand breakage repair by base excision repair method [16].
It was reported that gene expression profiles and somatic genetic changes of BRCA1 and BRCA2 related pancreatic cancer are different from sporadic cases. The histopathological and immunohistochemical characteristics of BRCA mutated patients shown poor prognosis. Despite these findings, conflicting data exist as to whether the prognosis of hereditary pancreatic cancer differs from that of sporadic cases. Some of the discrepancies may be explained by methodological differences or biases. However, no mutation-based studies have shown a survival advantage for BRCA1/2 mutation carriers and several unrelated studies have recently found that the presence of a BRCA1/2 mutation was an independent poor prognostic factor. Germ line mutations in the tumour suppressor genes breast cancer antigen gene BRCA1 and BRCA2 have been proven to portend a drastically increased lifetime risk of breast and ovarian cancers in the individuals who carry them. A number of studies have shown that the third most common cancer associated with these mutations is pancreatic cancer.
Several other lines of evidence also suggest that carriers of BRCA1 or BRCA2 mutations face an increased risk of pancreatic cancer [17,18]. In patients with sporadic pancreatic cancer, BRCA1/2 are mutated in the most advanced pancreatic intraepithelial neoplasia lesions, whereas a germ line mutation in either gene represents the earliest risk factor of patient’s close relatives. It has been reported that pancreatic cancer is the third most common cancer associated with BRCA1/2 mutations [19]. It was found that BRCA2 mutation poses an increased risk for developing pancreatic cancer [20]. Some study proposed BRCA2 as a genetic factor as causes of the pancreatic cancer [21]. As like breast cancer, 5-10% of pancreatic cancer cases are believed to be hereditary. Through analysis of the literature it was found that patients with pancreatic cancer and germ line BRCA2 mutations have a younger than average age of disease onset in case of Ashkenazi Jewish [22]. Satdler, et al. in 2012 established a strong family history of pancreatic cancer in a study among 211 Ashkenazi Jewish Proband. Among them 31% had a first-degree relative with pancreatic cancer, 53% had a second-degree relative and 16% had a third-degree relative diagnosed with the disease [23]. From the study it was established that BRCA1/2 mutations are most important factor to develop familial breast-pancreas cancer families and carriers of the BRCA2 mutation have an increased risk of developing pancreatic cancer. The use of different analysis model can be useful to establish variations in mutation prevalence. This review outlines the therapeutic approach to patients at high risk of developing pancreatic cancer, including criteria for genetic testing.
DNA damage
Different genotoxic agents vary in the type of DNA damage they inflict and the specificity of the induced damage triggers a variety of cellular responses specific to the type of lesion inflicted. While a large host of agents are known to activate checkpoint pathways, two commonly employed agents include IR and ultraviolent (UV) light. IR, by definition, is radiation with sufficient energy to ionize molecules with which it collides [24]. IR can damage DNA directly, or indirectly, through reactive oxygen species intermediates. IR is known to induce a large variety of DNA lesions, the most lethal of which is the DNA Double Stranded Break (DSB). The most lethal form of DNA damage is generally regarded to be the DSB. DSBs are generated endogenously, as a normal part of the cellular process, through replication fork collapse, during DNA replication and in repair events, and by exogenous agents such as ionizing radiation (IR) and other genotoxic compounds. Repair of DSBs is of cardinal importance in preventing chromosomal fragmentation, translocations and deletions. The genomic instability resulting from persistent or incorrectly repaired DSBs can lead to carcinogenesis through activation of oncogenes, inactivation of tumour-suppressor genes, or loss of heterozygosity (LOH) at specific loci, while in the germ line they can lead to inborn defects. The deleterious effects of DSBs have triggered the evolution of multiple pathways for their repair [25].
BRCA1 & BRCA2 function in the DNA damage response
The ability to precisely control the order and timing of cell cycle events is essential for maintaining genomic integrity and preventing mutations able to disrupt normal growth controls. Cells exposed to DNA damaging agents, such as ionizing radiation, coordinately arrest the progression of the cell cycle at the G1/S phase, the S phase and the G2/M phase to allow adequate time for damage repair [26]. It is now widely accepted that both BRCA1 and BRCA2 play multiple critical roles in the maintenance of genome stability as evidenced by a profound number of chromosomal translocations, duplications, and aberrant fusion events between non-homologous chromosomes in BRCA1 and BRCA2 deficient cells. BRCA1 plays a critical role in responding to DSBs through its function in HR. Firstly BRCA1 is recruited to DNA damage sites (Figure 1). Then BRCA1 recruits BRCA2, which facilitates Rad51 filament formation on the ssDNA [27]. Rad51 catalyzes the invasion of the homologous sequence on the sister chromatid, which is then used as template for accurate repair of the broken DNA ends. Other studies have shown that BRCA1 colocalizes with Rad50, a member of the MRN complex, following the induction of DNA damage; Mre11 encodes nuclease activity which resects flush ends of DSBs to generate ssDNA tracts. BRCA1 binds DNA directly and inhibits this Mre11 activity regulating the length and the persistence of ssDNA generation at sites of DNA damage. As ssDNA is a substrate for DNA repair by HR, it appears that BRCA1 might play an essential role in HR-mediated repair of DSBs through its inactivation of Mre11; an idea confirmed by the observation the HR is defective in BRCA1-deficient cells [28]. The roles played by BRCA1 and BRCA2 in the repair of DSBs by HR appear to differ, as evidence indicates a more direct role for BRCA2. The physical interaction between BRCA2 and Rad51 is essential for HR repair of DSBs to take place; BRCA2 is thought to be required for the transport of Rad51 from its site of synthesis to the site of DNA damage, where Rad51 is then released to form the nucleoprotein filament required for HR to take place [28].
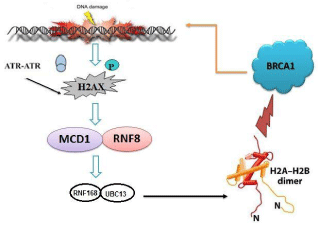
Figure 1: BRCA1 recruitment at the DNA damaged site.
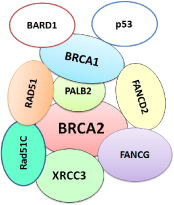
Figure 2: BRCA complex.
BRCA mutation effect
Individuals having BRCA1/2 germ line mutation are very potent to develop breast and ovarian cancer. This two gene mutation can lead to develop another type of cancer called pancreatic cancer [17,18]. Carriers of BRCA1/2 mutated genes are also very potent to develop this cancer as this genes are autosomal dominant in nature. Sporadic pancreatic cancer, having BRCA1/2 mutation can develop the most advanced pancreatic intraepithelial neoplasia lesions, in gremlin mutation it is hereditary. Pancreatic cancer is the third most cancer in patients who are having BRCA1/2 mutation [19].
A study among 211 Ashkenazi Jewish Proband. Within the sample, 31% had a first-degree relative with pancreatic cancer, 53% had a second-degree relative and 16% had a third-degree relative diagnosed with the disease. They are affected because of BRCA1/2 mutation [29]. The use of different analysis models within studies showed that pancreatic cancer has been regarded as a component of the breast-ovarian cancer syndrome.
Carriers of the BRCA2 mutation have an increased risk of developing pancreatic cancer [29]. It is clear that how the degree to which family history of pancreatic cancer influences the likelihood of detecting a BRCA1/2 mutation in an individual with breast cancer. Pancreatic cancer has been considered as a component of the breastovarian cancer syndrome. The utilization of different analysis models within studies can lead to variations in mutation prevalence.
In a study by Axilbund, et al. [30] over half the study population reported a family history of breast and/or ovarian cancer in addition to pancreatic cancer. The study findings suggest that BRCA1 mutations are not a substantial cause of breast cancer in familial pancreatic cancer kinships as none of the participants were found to possess a BRCA1 mutation from DNA sequencing.
Stadler, et al. [29] proved that 70 families had more than two relatives identified a disease with breast cancer within the same family members as the family history of related to the pancreas cancer and 31 probands had a relative with ovarian cancer within the same family tree. They also identified 14.2% BRCA1/2 sequence variation among the Ashkenazi Jews who were reported a personal history of breast cancer and a family history of pancreas cancer. Axilbund, et al. [30] showed that among 66 pancreas cancer patients, four having breast cancer before identified with pancreas cancer. Looking at things in the opposite way, in a study by Tulinius, et al. [31] it was found that, no family-related risk due to BRCA2 mutation was found for pancreas cancer among breast cancer patients, yet, it was obvious for cancers of the stomach, prostate and kidneys. These results could suggest that particular areas of BRCA1/2 mutation may have increased associations with particular cancers.
Kim, et al. [32] reviewed 1312 family pedigrees tested for BRCA1/2 mutations. They found, 219 families were positive for BRCA1 changes and 156 families had BRCA2 changes. Outcomes of the study showed that 11% of the 219 BRCA1 positive families had at least one relative with pancreatic cancer and 2.7% had more than one relative with pancreatic cancer. What’s more, Bermejo and Hemminki [33] shown that families of patients with breast cancer are before 35 years of age, presented significant incidence ratios related to the pancreatic cancers. However, this standard ratio was reported to be a strong sign of some association of early-beginning breast cancer and pancreatic cancer through causes unrelated to BRCA changes. But no other proposed causes were suggested also.
Observations of literature taken to be a number of studies being part are being made clear of mutated BRCA1/2 genes were responsible for an important fraction of pancreatic cancer development, as well as sensitivity to increased disease. Confirmed associations of BRCA1/2 changes with pancreatic cancer at a population level have been reported within the research. Holter, et al. [34] did not discover a statistically important connection of BRCA-mutation position with personal history of cancer. Increasing facts supporting across cancer with changes in structure in the BRCA genes has suggested that these tumors have nothing like it feeblenesses to special DNA-damaging agents and DNA repair inhibitors [35]. However how best to make out and pleasure these persons getting care remains a physical acts offer. Increasing evidence across malignancies with mutations in the BRCA genes has suggested that these tumors have unique susceptibility to specific DNA-damaging agents and DNA repair inhibitors [35]. However identification and treatment of these patients still remains a challenge.
Other important genes
BRCA1-associated protein 1 (BAP1) enzyme binds to the breast cancer type 1 susceptibility protein (BRCA1) via its RING finger domain [36]. Its involved with the regulation of DNA transcription, the cell cycle, cellular growth, response to DNA damage, and chromatin dynamics including chromatin remodelling [37]. It has been considered a tumor suppressor gene. Mutation on this gene also associated with many cancers. Luchini, et al. [38] in 2016 describe the BAP1 mutation influence the prognosis of cancer and it’s strongly associated with high-grade of cancer. This gene also responsible for the cancer-specific mortality as well as recurrence of cancer. They find BAP1 mutated cancer is more common in women than in men. The mutation can be detected by genomic analysis by AR MS-PCR, DNA sequencing and also immunohistochemistry (IHC) staining as the protein is normally expressed in nuclei, so the lack of expression can be consider as an abnormal BAP1 expression like mutation. The IHC methods can be useful to decide which high-risk patients should be screened first for the BAP1 gene mutation.
PALB2 (Partner and Localizer of BRCA2) binds to and colocalizes with BRCA2 in DNA repair. PALB2 is considering also Fanconi anemia (FA) genes that function in the FA-Breast Cancer (BRCA). The PALB2 gene is a tumor suppressor gene that interacts with both BRCA1 and BRCA2 during double-strand DNA repair. PALB2 acts as a physical link between BRCA1 and BRCA2 to form a “BRCA complex” the complex shown in Figure 2. Jones, et al. in 2009 analysed 100 families with familial pancreatic cancer and found four families with protein-truncating mutations in PALB2 [39]. Tischkowitz, et al. also confirmed the role of PALB2 mutation in pancreatic cancer after analysis of 21 cases with a family history of pancreatic cancer [40]. No specific screening guidelines for pancreatic cancer to detect PALB2 are available till now. Exact risk for pancreatic cancer conferred by PALB2 mutations yet not also been established. Recently it was found that PALB2 mutations occur with a prevalence of 2.1% in a population of BRCA1/2-negative breast cancer patients specifically selected for a personal and/or family history of pancreatic cancer [41].
ARID1 gene is another important gene which considered as a tumor suppressor gene. It is located in chromosome 1p36.11. Deletion or any mutation of this gene is responsible to develop many kind of cancer, especially in those arising from ectopic or eutopic endometrium. ARID1A, play the role as a “gatekeeper”, and “caretaker” as it maintains the genomic stability by preventing sequence mutations and structural aberrations in chromosomes [42]. ARID1 genes interact with SWI/SNF complexes and play an important role chromosome remodelling. Its regulates gene expression by controlling gene accessibility [43]. Chandler, et al. demonstrated that ARID-DNA interactions promotes to SWI/SNF activity in mouse embryos. If the mutation presents in this gene the ARID1A-DNA interactions became disrupted. This results in a decrease in promoter occupancy by SWI/SNF [44]. Mutation in ARID1B gene is associated with pancreatic cancer and other cancer [45]. Luchini, et al. [46] demonstrated that ARID1A loss promotes cancer-specific mortality, and also help to recurrence of cancer. They conclude that this gene should be considered as an important potential target for personalized medicine in cancer treatment. Some research suggested that ARID1A-mutated cancer may also be treated by targeting residual SWI/SNF activity, the PI3K/AKT pathway, the tumor immunological microenvironment, stabilizing wild-type p53 and by targeting the DNA damage response [47]. Recently it has been found that mutated ARID1A also associated with defects in DNA repair [48]. Such mutations can be another possible target of PARPi to treat cancer.
Targeted therapies for BRCA mutation carrier’s
DNA damage response genes play vital roles in the maintenance of genome stability. Defects in cell cycle checkpoint and DNA repair genes, especially mutation, are associated with a wide spectrum of Cancer. Several genes that participate in the DNA damage response. List of DDR genes is given in Table 1. Many of these genes act as a regulator of other genes implicated in the pathogenesis of cancer. DNA damage response (DDR) and tumor evolution will help to understand the mechanisms of BRCA associated tumorigenesis, as well as the development of therapeutic approaches. Sequencing of panels of relevant genes for clinical use, as well as whole exome and whole genome sequencing for research purposes become increases now as because the genomic sequencing technologies have drastically decreased in cost. It is likely that eventually all patients will have their tumors and matched germ line sequenced. In addition to directly identifying patients with germ line and somatic mutations in well-known DDR genes, it may be possible to identify patients with tumors that have a BRCAness phenotype. Mutational profile can be established using a whole-exome sequencing approach [49]. A diagnostic company announced approval from the U.S. FDA on the same day for their BRAC Analysis CDx diagnostic kit (https:// www.myriadpro.com/) to be used as the only companion diagnostic in conjunction with olaparib. BRAC Analysis CDx is this company’s first FDA-approved companion diagnostic for use with a novel PARP inhibitor. It is a highly accurate molecular companion diagnostic test that identifies deleterious or suspected deleterious mutations in BRCA1 and BRCA2 genes using DNA obtained from a blood sample.
![]()
Gene Name
Chromosome location
Base excision repair genes
UNG
12q24.11
SMUG1
12q13.13
MBD4
3q21.3
TDG
12q23.3
OGG1
3p25.3
MUTYH
1p34.1
NTHL1 (NTH1)
16p13.3
MPG
16p13.3
NEIL1
15q24.2
NEIL2
8p23.1
NEIL3
4q34
APEX1
14q11.2
APEX2
Xp11.21
LIG3
17q12
XRCC1
19q13.31
PNKP
19q13.33
APLF (C2ORF13)
2p13.3
PARP1
1q42.12
PARP2
14q11.2
PARP3
3p21.1
MGMT
10q26.3
ALKBH2 (ABH2)
12q24.11
ALKBH3 (DEPC1)
11p11.2
TDP1
14q32.11
TDP2 (TTRAP)
6p22.3
MSH2
2p21
MSH3
5q14.1
MSH6
2p16.3
MLH1
3p22.3
PMS2
7p22.1
MSH4
1p31.1
MSH5
6p21.33
MLH3
14q24.3
PMS1
2q32.2
PMS2L3
7q11.23
XPC
3p25.1
RAD23B
9q31.2
CETN2
Xq28
RAD23A
19p13.13
XPA
9q22.33
DDB1
11q12.2
DDB2 (XPE)
11p11.2
RPA1
17p13.3
RPA2
1p35.3
RPA3
7p21.3
ERCC3 (XPB)
2q14.3
ERCC2 (XPD)
19q13.32
GTF2H1
11p15.1
GTF2H2
5q13.2
GTF2H3
12q24.31
GTF2H4
6p21.33
GTF2H5 (TTDA)
6p25.3
CDK7
5q13.2
CCNH
5q14.3
MNAT1
14q23.1
ERCC5 (XPG)
13q33.1
ERCC1
19q13.32
ERCC4 (XPF)
16p13.12
LIG1
19q13.32
ERCC8 (CSA)
5q12.1
ERCC6 (CSB)
10q11.23
UVSSA (KIAA1530)
4p16.3
XAB2 (HCNP)
19p13.2
MMS19
10q24.1
RAD51
15q15.1
RAD51B
14q24.1
RAD51D
17q12
DMC1
22q13.1
XRCC2
7q36.1
XRCC3
14q32.33
RAD52
12p13.33
RAD54L
1p34.1
RAD54B
8q22.1
BRCA1
17q21.31
SHFM1
7q21.3
RAD50
5q23.3
MRE11A
11q21
NBN (NBS1)
8q21.3
RBBP8 (CtIP)
18q11.2
MUS81
11q13.1
EME1
17q21.33
EME2
16p13.3
GIYD1
16p11.2
GIYD2
16p11.2
GEN1
2p24.2
FANCA
16q24.3
FANCB
Xp22.31
FANCC
9q22.32
BRCA2
13q13.1
FANCD2
3p25.3
FANCE
6p21.31
FANCF
11p14.3
FANCG
9p13.3
FANCI
15q26.1
BRIP1
17q23
FANCL
2p16.1
FANCM
14q21.3
PALB2
16p12.1
RAD51C
17q23.2
BTBD12
16p13.3
FAAP20
1p36.33
FAAP24
19q13.11
XRCC6
22q13.2
XRCC5
2q35
PRKDC
8q11.21
LIG4
13q33.3
XRCC4
5q14.2
DCLRE1C
10p13
NHEJ1
2q35
NUDT1
7p22.3
DUT
15q21.1
RRM2B
8q22.3
POLB
8p11.21
POLG
15q26.1
POLD1
19q13.33
POLE
12q24.33
PCNA
20p12.3
REV3L
6q21
MAD2L2
1p36.22
REV1L
2q11.2
POLH
6p21.1
POLI
18q21.2
POLQ
3q13.33
POLK
5q13.3
POLL
10q24.32
POLM
7p13
POLN
4p16.3
FEN1
11q12.2
FAN1
15q13.2
TREX1
3p21.31
TREX2
Xq28
EXO1
1q43
APTX
9p21.1
SPO11
20q13.32
ENDOV
17q25.3
Ubiquitination and modification
UBE2A
Xq24-q25
UBE2B
5q31.1
RAD18
3p25.3
SHPRH
6q24.3
HLTF
3q25.1-q26.1
RNF168
3q29
SPRTN
1q42.12-q43
RNF8
6p21
RNF4
4p16.3
UBE2V2
8q11.21
UBE2N
12q22
H2AFX
11q23.3
CHAF1A
19p13.3
SETMAR
3p26
Genes defective in diseases associated with sensitivity to DNA damaging agents
BLM
15q26.1
WRN
8q24.3
ATM
11q22.3
TTDN1
7p14
Table 1: Genes responsible for DNA damage Response (DDR).
Mafficini, et al. in 2016 [50] describe the somatic and germ line mutations detection of BRCA1 and BRCA2 using formalinfixed paraffin-embedded (FFPE) tissues. They use next-generation sequencing (NGS) to detection the variation in those genes. They successfully established that the next generation sequencing performed with a commercial kit (HR1, 4 Bases) is suitable for not only germ line but also somatic mutations in these genes [49]. This method may be the tool of choice for the early detection of cancer due to its ability to perform massively parallel DNA sequencing. However this protocol has a limitation on single molecule tagging, low sensitivity and cannot be useful for low quality DNA sequencing. Due to the high costing, it is not feasible to implement this method in low or middle income country.
Waddell, et al. [51] recently published a study in which whole genome sequencing and copy number variation analysis was done on 100 prospectively collected pancreatic cancer specimens. This group reported a roughly similar prevalence of germ line BRCA mutations as Holter, et al [34], and they in addition found germ line and somatic mutations in eight genes in DNA-damage repair pathways (including BRCA1/2). Importantly, they observed that in case of 14% of patients having BRCA mutation, were more likely to have an unstable pattern of genomic structural variation. They proven that the enormous overlap among the mutation status (germ line and somatic) of the eight recognized BRCA-pathway genes, the genomically unstable subtype, and a previously defined BRCA-mutational signature [52]. In addition they showed that the tumors with those DDR deficits had been related to platinum based therapy. These findings suggest that there may be a class of sporadic pancreatic cancer that share a soreferred to as BRCAness phenotype with germ line mutant tumors, arising from DNA repair defects due to a compromised DNA restore via homologous recombination [53]. These outcomes suggest that it is able to be feasible to use genomic biomarkers of defective DNA maintenance to identify a larger group of patients with BRCAness that would advantage from remedies targeting DDR pathways.
Targeted therapy based on inhibiting the DNA damage response (DDR) is a greater therapeutic strategy to treat patients with tumors lacking specific DDR functions. Mark J. O’Connor in 2015 describes the different concepts behind targeting DDR in cancer and the significant opportunities for DDR-based therapies. The most prominent example is the invention of poly (ADP-ribose) polymerase (PARP) inhibitors on BRCA1- or BRCA2-defective tumors, which takes advantage of the defects in DNA repair in cancer cells. [54]. Holter, et al [34] describe their systematic investigation of the prevalence rates of infective germ line mutations in an exceedingly range of DNA damage response (DDR) genes in patients with pancreatic cancer. Together with a lot of research on prevalence rates, the studies conjointly offer insight into clinical predictors which will be most relevant for determinant those patients to check for these mutations. Testing for BRCA1 and BRCA2 mutations in breast and ovarian cancer has become routine in those deemed as high risk by virtue of case history. Whereas patients with pancreatic cancer with BRCA mutations measure smaller in range, these patients also are in an exceedingly position to benefit from treatment. These treatments are the platinum-based chemotherapy agents and the newer class of drugs known as poly (ADP-ribose) polymerase (PARP) inhibitors [35]. PARP inhibition leads to transformation of single-strand breaks into double-strand breaks that are cytotoxic in cells and renders them unable to be repaired through homologous repair. BRCA mutated cells of pancreatic cell lines are sensitive to cisplatin, mitomycin, and PARP inhibitors. While DNA defects are often a step in the process of tumourigenesis, once a cell becomes cancerous, such defects may be exploitable to enhance susceptibility to chemotherapeutic agents. It is well accepted that BRCA1 deficiency leads to the dysregulation of DNA repair pathways, which in turn renders tumour cells more vulnerable to DNA damaging agents. The PARP family of enzymes serves a vital role in the repair of single-stranded DNA breaks (SSBs). Normally, unrepaired SSBs lead to double-strand breaks (DSBs), which are subsequently repaired in cells with normal BRCA function. However, in cells where BRCA is nonfunctioning or deficient, DSB are left unrepaired, leading to genomic instability and cell death).
Identification of patients with mutations in DDR genes is becomes a great interest to evaluate whether or not these can respond higher to targeted treatment methods. Research and development in this area have faced significant challenges to lack of physician awareness on such cancer association with BRCA1/2 mutations, small incidence rate, and no standard methodology to selection of patients for genetic testing, disease aggressiveness progression that limits opportunity for genetic testing. Olaparib is an oral poly (ADP-ribose) polymerase inhibitor with activity in germ line BRCA1 and BRCA2 (BRCA1/2) associated cancer. Some clinical trial shows interest to establish the role of olaparib for the treatment of ovarian and pancreatic cancer in BRCA-mutation carriers. Kaye, et al. in 2012 suggested that 400 mg twice per day of Olaparib is a suitable for the patents [55].
Veliparib is a potent oral irreversible small molecule inhibitor of PARP 1 and 2 which are evaluated in non-randomized trials in combination with cisplatin-based therapy in previously treated and also untreated patients with known BRCA or PALB2 mutations [56]. They observed that very high level of activity within the patients. Their survival rate also very impressive, whereas no significant activity was observed in non-BRCA-mutated patients. This is the first prospective randomized trial of this combination is underway in BRCA/PALB2- mutated patients with stage III/IV disease.
Conclusion
If a biomarker for DDR-deficient tumors was validated based on the genomic scar or mutational footprint, tremendous quantity of sufferers is probably identified to enroll in medical trials of particular platinum marketers and/or PARP inhibitors, in addition to some of different small molecules in improvement focused on DDR pathways. Because the scientific use of genomic sequencing turns into more useful and hence there will be a great deal to study concerning differential sensitivities of diverse DDR mutations, which include somatic as well as germ line mutations, effects of mutations in one of a kind contributors of the same pathways, effects of cooperating mutations, and new mechanisms of treatment resistance. For now, however, clinical trials have focused on a narrow subset of patients with germ line BRCA1/2 mutations. Screening of patients with PDAC for germ line BRCA1/2 mutations could increase clinical trial enrolment for this devastating disorder that has been devoid of personalized therapeutic opportunities. The importance of BRCA1/2 mutation status, provide genetic counselling and are capable of imposing new treatment strategy for relevant cancers cannot be stressed enough.
References
- SEER Program: SEER Cancer Statistics Review 1975-2011.
- Lal G, Liu G, Schmocker B, Kaurah P, Ozcelik H, Narod SA, et al. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000; 60: 409-416.
- Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994; 8: 27-32.
- Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009; 324: 217.
- Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994; 54: 3025-3033.
- Ghiorzo P, Fornarini G, Sciallero S, Battistuzzi L, Belli F, Bernard L, et al. CDKN2A is the main susceptibility gene in Italian pancreatic cancer families. J Med Genet. 2012; 49: 164-170.
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996; 271: 350-353.
- Sato N, Rosty C, Jansen M, Fukushima N, Ueki T, Yeo CJ, et al. STK11/LKB1 Peutz-Jeghers gene inactivation in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol. 2001; 159: 2017-2022.
- Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012; 2: 41-46.
- Win AK, Young JP, Lindor NM, Tucker KM, Ahnen DJ, Young GP, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012; 30: 958-964.
- Fernandez E, La Vecchia C, D'Avanzo B, Negri E, Franceschi S. Family history and the risk of liver, gallbladder, and pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 1994; 3: 209-212.
- Klein AP, Hruban RH, Brune KA, Petersen GM, Goggins M. Familial pancreatic cancer. Cancer J. 2001; 7: 266-273.
- Bartsch DK, Kress R, Sina-Frey M, Grützmann R, Gerdes B, Pilarsky C, et al. Prevalence of familial pancreatic cancer in Germany. Int J Cancer. 2004; 110: 902-906.
- Thull DL, Vogel VG. Recognition and management of hereditary breast cancer syndromes. Oncologist. 2004; 9: 13-24.
- Knudson AG. Antioncogenes and human cancer. Proc Natl Acad Sci U S A. 1993; 90: 10914-10921.
- Prat J, Ribé A, Gallardo A. Hereditary ovarian cancer. Hum Pathol. 2005; 36: 861-870.
- Thompson D, Easton DF; Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002; 94: 1358-1365.
- Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst. 2006; 98: 1694-1706.
- Greer JB, Whitcomb DC. Role of BRCA1 and BRCA2 mutations in pancreatic cancer. Gut. 2007; 56: 601-605.
- Stadler ZK, Salo-Mullen E, Patil SM, Pietanza MC, Vijai J, Saloustros E, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012; 118: 493-499.
- Lucas AL, Frado LE, Hwang C, Kumar S, Khanna LG, Levinson EJ, et al. BRCA1 and BRCA2 germline mutations are frequently demonstrated in both high-risk pancreatic cancer screening and pancreatic cancer cohorts. Cancer. 2014; 120:1960-1967.
- Naderi A, Couch FJ. BRCA2 and pancreatic cancer. Int J Gastrointest Cancer. 2002; 31: 99-106.
- Stadler ZK, Salo-Mullen E, Patil SM, Pietanza MC, Vijai J, Saloustros E, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012; 118: 493-499.
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press. Washington. DC. USA. 2006.
- Ivanov EL, Haber JE. DNA repair: RAD alert. Curr Biol. 1997; 7: R492-495.
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004; 432: 316-323.
- Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005; 3: 531-539.
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999; 4: 511-518.
- Stadler ZK, Salo-Mullen E, Patil SM, Pietanza MC, Vijai J, Saloustros E, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012; 118: 493-499.
- Axilbund JE, Argani P, Kamiyama M, Palmisano E, Raben M, Borges M, et al. Absence of germline BRCA1 mutations in familial pancreatic cancer patients. Cancer Biol Ther. 2009; 8: 131-135.
- Tulinius H, Olafsdottir GH, Sigvaldason H, Arason A, Barkardottir RB, Egilsson V, et al. The effect of a single BRCA2 mutation on cancer in Iceland. J Med Genet. 2002; 39: 457-462.
- Kim DH, Crawford B, Ziegler J, Beattie MS. Prevalence and characteristics of pancreatic cancer in families with BRCA1 and BRCA2 mutations. Fam Cancer. 2009; 8: 153-158.
- Lorenzo Bermejo J, Hemminki K. Risk of cancer at sites other than the breast in Swedish families eligible for BRCA1 or BRCA2 mutation testing. Ann Oncol. 2004; 15: 1834-1841.
- Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. 2015; 33: 3124-3129.
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012; 481: 287-294.
- Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, et al. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009; 69: 111-119.
- Eletr ZM, Wilkinson KD. An emerging model for BAP1's role in regulating cell cycle progression. Cell Biochem Biophys. 2011; 60: 3-11.
- Luchini C, Veronese N, Yachida S, Cheng L, Nottegar A, Stubbs B, et al. Different prognostic roles of tumor suppressor gene BAP1 in cancer: A systematic review with meta-analysis. Genes Chromosomes Cancer. 2016; 55: 741-749.
- Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009; 324: 217.
- Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N, et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009; 137: 1183-1186.
- Hofstatter EW, Domchek SM, Miron A, Garber J, Wang M, Componeschi K, et al. PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer. 2011; 10: 225-231.
- Wu RC, Wang TL, Shih IeM. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014; 15: 655-664.
- Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013; 3: 35-43.
- Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, Magnuson T. ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol Cell Biol. 2013; 33: 265-280.
- Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A. 2012; 109: E252-259.
- Luchini C, Veronese N, Solmi M, Cho H, Kim JH, Chou A. Prognostic role and implications of mutation status of tumor suppressor gene ARID1A in cancer: a systematic review and meta-analysis. Oncotarget. 2015; 6: 39088-39097.
- Bitler BG, Fatkhutdinov N, Zhang R. Potential therapeutic targets in ARID1A-mutated cancers. Expert Opin Ther Targets. 2015; 19: 1419-1422.
- Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015; 5: 752-767.
- Wu J, Jiao Y, Dal Molin M, Maitra A, Roeland F. de Wilde, Laura D. Wood, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011; 108: 21188-21193.
- Mafficini A, Simbolo M, Parisi A, Rusev B, Luchini C, Cataldo I, et al. BRCA somatic and germline mutation detection in paraffin embedded ovarian cancers by next-generation sequencing. Oncotarget. 2016; 7: 1076-1083.
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015; 518: 495-501.
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500: 415-421.
- Turner N, Tutt A, Ashworth A. Hallmarks of 'BRCAness' in sporadic cancers. Nat Rev Cancer. 2004; 4: 814-819.
- Ashworth A. A synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008; 26: 3785-3790.
- Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012; 30: 372-379.
- O’Reilly EM. BRCA-mutated pancreas adenocarcinoma: Emerging therapeutic implications. AACR Special Conference on Pancreatic Cancer. Abstract IA28. 2014.