
Review Article
Austin J Gastroenterol. 2017; 4(2): 1081.
Paradoxical Inflammation in IBD Patients Treated with Anti-Tumor Necrosis Factor Alpha Therapy
Münger C¹, Juillerat P¹ and Niess JH1,2*
¹Department of Gastroenterology, University Clinic of Visceral Surgery and Medicine, Switzerland
²Department of Gastroenterology and Hepatology, University Hospital of Basel, Switzerland
*Corresponding author: Niess JH, Department of Gastroenterology and Hepatology, University Hospital Basel, Petersgraben 4, 4031 Basel, Switzerland
Received: February 07, 2017; Accepted: April 03, 2017; Published: May 03, 2017
Abstract
The development of tumor necrosis factor (TNF) antagonists has revolutionized the treatment of inflammatory bowel disease (IBD). Paradoxically, TNF antagonists can also induce inflammation, such as psoriasis induced by TNF antagonists. The mechanisms by which TNF antagonists induce paradoxical inflammation, has remained largely unresolved. In this review we will focus on the immunologic effects of the therapy with TNF antagonists and give an overview of TNF antagonist-induced immunologically mediated diseases.
Keywords: Ulcerative colitis; Crohn’s disease; Tumor necrosis factor; Paradoxical inflammation
Abbreviations
cIAP1: Cellular Inhibitor of Apoptosis Protein 1; cIAP2: Cellular Inhibitor of Apoptosis Protein 2; CKI: Intracellular Casein Kinase I; CRH: Corticotropin Releasing Hormone; EGFR: Epidermal Growth Factor Receptor; FcγRI: Fc-Gamma Receptor I; FcγRIII/II: Fc-Gamma Receptor III/II; FLIPL: FLICE-like Inhibitory Protein; G-CSF: Granulocyte Colony Stimulating Factor; HS: Hidradenitis Suppurativa; IKK: IκB Kinase; IBD: Inflammatory Bowel Disease; JNK: JUN N-terminal Kinase; LPS: Lipopolysaccharide; MLKL: Mixed Lineage Kinase Domain-Like (1); NF–κß: Nuclear Factor �Kappa-Light-Chain-Enhancer’ of Activated B-Cells; RIPK: Receptor- Interacting Serine/Threonine-Protein Kinase 1; sTNF: Soluble Tumor Necrosis Factor; TAB2: TGFß-Activated Kinase 2; TAB3: TGFß-Activated Kinase 3; TACE: TNF-Alpha-Converting Enzyme; tmTNF: Transmembrane Tumor Necrosis Factor; TNF: Tumor Necrosis Factor; TNFR1: Tumor Necrosis Factor Receptor 1; TNFR2: Tumor Necrosis Factor Receptor 2; TNFSF: Tumor Necrosis Factor Superfamily; TNFSRF: Tumor Necrosis Factor Receptor Superfamily; TRADD: TNFR1-Associated Death Domain Protein; TRAF2: TNFRAssociated Factor 2; TRAF5: TNFR-Associated Factor 5; UV: Ultraviolet
Introduction
The tumor necrosis factor (TNF) antagonists are potent therapies for patients with inflammatory bowel disease (IBD) and other immune-mediated diseases. TNF plays a key role in a complex network of cytokines in initiating inflammatory reactions by activation of NF- κB, induction of apoptosis and necroptosis [1-3]. Forward signaling is induced by binding of soluble TNF to the TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), whereas reverse signaling is induced by binding of the respective receptors to membrane-bound TNF [1,4]. This leads to pleiotropic effects, such as the release of the corticotropin releasing hormone (CRH) by the hypothalamus [5], suppression of appetite [6], release of acute phase proteins by the liver, such as C reactive protein, stimulation of phagocytes, attraction of neutrophils, increase of insulin resistance by serine-phosphorylation of insulin receptor substrate-1 in the liver an peripheral tissues [7-12]. Activation of TNF pathways can hence induce shock-like symptoms [12]. Prolonged exposition of low TNF concentrations can lead to cachexia as observed in cancer patients. Interfering with these complex processes is an attractive target to treat auto-inflammatory diseases, in which elevated TNF concentrations have been reported [13,14]. However, the pharmacological interference with TNF can also have unexpected effects. For example, the application of these substances can lead to immunologic side effects, either by the induction of antidrug antibodies, which can lead to a loss of response or infusion reactions or by a shift in the complex cytokine network which can paradoxically lead to other immune-mediated diseases such as psoriasis, arthritis and sarcoidosis that usually respond well to the treatment with TNF antagonists [15-18]. A nomenclature for the adverse events of biologicals analogue to the established nomenclature for adverse events induced by traditional drugs has been suggested (Table 1) [19]. We hence searched the literature using the key words “paradoxical reaction”, “IBD”, “ulcerative colitis”, “Crohn’s disease”, “anti-tumor necrosis factor-alpha”, “infliximab”, “adalimumab, “etanercept”, “golimumab”, “certolizumab”, “psoriasis”, “psoriatic arthritis”, “alopecia”, “arthritis”, “vasculitis”, “sarcoidosis”, and “hidradenitis suppurativa”. In this review we will first give an overview of the current concepts of TNF signaling pathways and then focus on paradoxical immunologic side effects of TNF antagonists.
![]()
Type of reaction
Mechanism
Example
Type a reaction
Reactions related to cytokine and cytokine release syndrome
Sweet’s syndrome induced by granulocyte colony stimulating factor (G-CSF)
Type β reaction
Hypersensitivity reaction linked to the immunogenicity of biological agents
Loss of efficacy due to the development of anti-drug antibodies
Type γ reaction
Related to immunodeviation
Immunodeficiency, cancer risk, new-onset autoimmune disease
Type d reaction
Related to the co-expression of the target antigen on both pathologic and normal tissue
Acneiform eruptions induced by epidermal growth factor receptor (EGFR) inhibitor
Type e reaction
New and unexpected physiological functions of biological agents revealed by the in vivo use in humans
Aggravation of heart failure by TNF antagonists
Table 1: Classification of adverse side effects of biological agents.
What is TNF and why is it an important drug target?
TNF is one of the best known members of the tumor necrosis factor superfamily (TNFSF), which comprises 19 ligands involved in pathways leading to cell survival, proliferation, differentiation, apoptosis or necroptosis. To date 29 members of the TNF receptor superfamily (TNFRSF) have been identified [2]. TNF was first described in 1975 as a protein that induces tumor cell death and named hence this protein tumor necrosis factor [20]. Later in the early eighties TNF was cloned and purified, and was shown to be identical with the murine protein cachectin, a macrophage-derived protein responsible for the wasting syndrome in chronic infected animals (cachexia) [3]. Later its role in inflammatory processes but also in cellular communication, cell differentiation and cell death has been recognized.
TNF plays a key role in the cytokine network of inflammatory reactions. Its expression can initiate inflammatory reaction, but can also have anti-inflammatory effects that might be important in the resolution of inflammatory reactions, depending on the cell types involved and the local tissue milieu [4]. One hypothesis for the explanation of the anti-inflammatory activity of TNF suggests that TNF has the capacity to induce apoptosis of immune cells responsible for inflammation [21]. TNF is also a critical factor of local glucocorticoid synthesis in the intestinal mucosa during the course of experimental colitis in mice. The absence of TNF in experimental models leads to an exacerbation of disease [22]. Thus, the proinflammatory action of TNF seems to be critical for the initiation of inflammation, whereas its anti-inflammatory function helps to resolve the inflammation.
TNF is a pleiotropic protein; it mediates a wide variety of biologic activities. Its different forms, the several possible interactions with receptors and the variety of signaling pathways create a complex biological system [4]. TNF is produced by macrophages, T cells, mast cells, granulocytes, natural killer (NK) cells, fibroblasts, neurons, keratinocytes and smooth muscle cells [4]. TNF exists as a transmembrane receptor (tmTNF), from which the soluble factor (sTNF) can be released after being enzymatically cleaved by the metalloproteinase TNF-converting enzyme (TACE; also known as ADAM17) [23]. Both sTNF and tmTNF interact with TNF receptor 1 (TNFR1) and TNF receptor 2 (TNRF2) [4]. TNFR1 is constitutively expressed on most nucleated cells, whereas only immune cells and endothelial cells almost exclusively express TNFR2. Binding of TNF to TNFR1 and TNFR2 leads to the activation of NF-κB. However, the signaling cascades of the respective receptors are different. The expression of TNF by different cell types, the different expression patterns of the respective receptors and several possibilities of interactions between sTNF and tmTNF with the respective receptors, helps to regulate a fine-tuned cell-specific immune response.
What signaling pathways are induced by TNF?
Before we discuss in detail the signaling cascades induced by TNF we will stress potential mechanisms by which TNF induces intracellular signaling pathways. Forward signaling (binding of sTNF or tmTNF to TNFR1 or TNFR2 on an effector cell leads to intracellular signaling in the effector cell) has to be distinguished from reverse signaling (tmTNF activates an intracellular signaling cascade in the tmTNF-bearing cell itself). Depending on the TNF receptor type, the presence and modification of intracellular proteins TNF signaling can lead to different biological outcomes. TNF signaling can ultimately result in (i) NF-κB activation leading to pro-inflammatory cytokine production (ii) apotosis or (iii) necroptosis. Necroptosis is a proinflammatory form of cell death, in which in contrast to apoptosis the initiating events are caspase-independent. Necroptotic cells display cell swelling, rupture of the cell membrane and uncontrolled release of cell material into the environment [24].
NF-κB activation: We will first focus on the cascade activated by binding of TNF to TNFR1 leading to NF-κB activation. Both TNFR1 and TNFR2 have cysteine rich extracellular domains but their intracellular domains are distinct. TNFR1 contains a cytoplasmic death domain to which the adaptor molecule TNFR1-associated death domain protein (TRADD) binds [25,26]. The TNFR1/TRADD recruits the receptor-interacting serine/threonine-protein kinase 1 (RIPK1), the TNFR-associated factor 2 (TRAF2), the TNFRassociated factor 5 (TRAF5) and the cellular inhibitor of apoptosis protein 1 (cIAP1) and the cellular inhibitor of apoptosis protein 2 (cIAP2) to form the TNFR1 complex I [27-29]. cIAP1 and cIAP2 are E3 ubiquitin ligases, which add polyubiquitin chains to the receptorinteracting serine/threonine-protein kinase 1 (RIPK1) [30,31]. Polyubiquitylated RIPK1 recruits MAP3K7-binding protein 2 (TAB2) and TAB3 and TGFß-activated kinase 1 (TAK1), which activates JUN N-terminal kinase (JNK), p38 and the IκB kinase (IKK) complex [32- 34]. IKK activates NF-κB, ultimately leading to the transcription of pro-inflammatory cytokines and anti-apoptotic factors (Figure 1), such as the long iso form of FLICE-like inhibitory protein (FLIPL) and BCL-XL to promote cell survival [35].
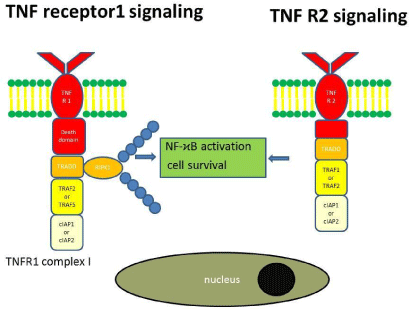
Figure 1: TNF receptor 1 and TNF receptor 2 signaling pathways.
By contrast to TNFR1 TNFR2 lacks a cytoplasmic death domain. After binding TNF TNFR2 recruits TRAF1 and TRAF2, which in turn ubiquitylates RIPK1 (K63-ubiliquation) to activate the IKK complex [36].
Apoptosis: Binding of TNF to TNFR1 can also result in apoptosis (programmed cell death). Downstream of TNFR1 RIPK1 is the central switch, which turns either the NF-κB activation or apoptosis pathway on or off [30]. When RIPK1 is ubiquitylated, the NF-κB activation pathway is activated. When RIPK1 is deubiquitylated or when ubiquitylation of RIPK1 is prevented, then the apoptosis initiating pathway is turned on [30]. Ubiquitin-modifying enzymes, such as A20, and deubiquitylating enzymes, such as Cezanne and cylindromatosis (CYLD) can remove ubiquitin chains from RIPK1 [37,38]. Deubiquitylated RIPK1 dissociates from the membrane, moves to the cytosol and interacts with TNFR1-associated TRADD to form the cytosolic complex IIa, which activates pro-caspase 8 and induces apoptosis [39]. In an alternative pathway cIAP1 and cIAP2 are removed preventing RIPK1 ubiquitylation, which dissociates from the membrane to assemble with FADD, pro-caspase 8, FLIPL and RIPK3 (but not TRADD) to form the complex IIb (also known as ripoptosome). This leads to the release of active caspase 8. There is hence a TRADD-dependent and TRADD-independent pathway by which TNF signaling induces classical apoptosis [40]. Intracellular signaling cascades that adapt to the respective tissue context and situation tightly control TNF signaling. RIPK1 is one of the essential switches of TNF signaling. When RIPK1 is poly-ubiquitylated the IKK complex is activated to induce NF-κB production, which binds to promoter regions required for the transcription of anti-apopotic factors. Thus, ubiquitylation of RIPK1 prevents death induction. When RIPK1 is not ubiquitylated, TNF-driven NF-κB expression is turned off and TNF-driven apoptotic signaling is turned on (Figure 2).
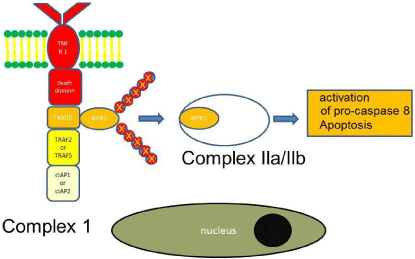
Figure 2: TNF signaling leading to apoptosis.
Necroptosis: Necroptosis is a pro-inflammatory cell death in which swelling of the organelles, rupture of the cell membrane and uncontrolled release of intracellular contents into the surrounding tissue occurs. In contrast to apoptosis, necroptosis is caspaseindependent. As for apoptosis deubiquitylated or non-ubiquitylated RIPK1 moves to the cytosol [41,42]. Then RIPK1 recruits several RIPK3s. RIPK1 and RIPK3 are not cleaved to prevent the activation of caspase. Instead RIPK1 and RIPK3 form microfilaments, which is called the necrosome. Together with mixed lineage kinase domainlike (MLKL) the necrosome induces necroptosis (Figure 3).
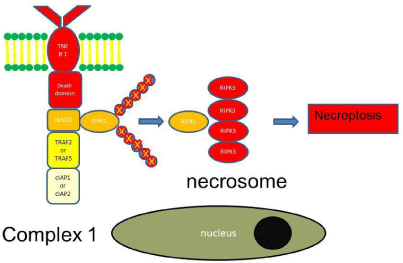
Figure 3: TNF signaling leading to necroptosis.
What is reverse signaling induced by TNF?
Reverse signaling is defined as a process in which tmTNF induces an intracellular signaling cascade in the cell expressing tmTNF itself [1]. Reverse signaling can occur after binding of membrane bound TNFRs, soluble TNFRs or antibodies to tmTNF. Reverse signaling is not specific to tmTNF. Other members of the TNF family, such as CD40L, LIGHT, TRANCE, TRAIL, CD30L, FasL, 4-1BBL, OX40L, CD70, can also induce reverse signaling. The detailed signaling cascade has not yet been uncovered, but members of the TNF family have an intracellular casein kinase I (CKI) binding site, which could mediate the signaling cascade [43]. tmTNF induced reverse signaling has cell-dependent effects. It increases the expression of adhesions molecules such as selectins, enhances cytotoxic effects of CD8 T cells and NK cells, induces proliferation of CD4 and CD8 T cells and modulates monocyte/ macrophage function. Reverse signaling downregulates the production of IL-1, TNF and IL-6 in macrophages pre-sensitized with lipopolysaccharide (LPS). This means that repeated challenges of macrophages does not lead to a continuous production of pro-inflammatory cytokines. Instead, the production of pro-inflammatory cytokines is down regulated, which is called LPS tolerance of macrophages [44].
What role does TNF play in inflammatory bowel disease?
TNF is involved in the pathogenesis of immune-mediated diseases, such as rheumatoid arthritis and Inflammatory bowel diseases (IBD). It is therefore an important target for the treatment of these diseases. In IBD, its important role in the pathogenesis of the disease has been recognized for a long time. Already more than 20 years ago, it has been shown that TNF levels in sera are elevated in diseased individuals as compared to healthy controls [14]. Advances of endoscopic technologies allow nowadays the identification of mucosal immunocytes that express membrane TNF [45]. Isolated mononuclear cells from the lamina propria of ulcerative colitis and Crohn’s disease patients are characterized by increased TNF production [46]. TNF regulates proliferation, apoptosis and necroptosis of intestinal epithelial cells, which may contribute to the disruption of the epithelial barrier leading to increased permeability of the barrier for luminal pathogens [47]. The importance of TNF in IBD is further highlighted by the success of antagonizing TNF by TNF antagonists, and the response to TNF antagonists directly correlates to the number of mucosal immunocytes expressing membrane TNFalpha before treatment is initiated [45].
How are TNF antagonists used in the treatment of patients with inflammatory bowel disease?
There are four TNF antagonists on the market for the treatment of IBD (Figure 4): Infliximab, a chimeric protein, and the fully humanized substances adalimumab, certolizumab pegol and golimumab. Etanercept, another TNF antagonist which binds soluble TNF has no effect in IBD [48].
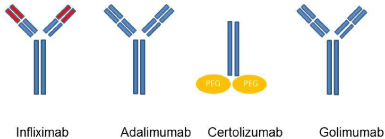
Figure 4: Simplified structure of the TNF antagonists.
Infliximab was the first TNF antagonist used in patients with Crohn’s disease and ulcerative colitis. Its efficacy was first demonstrated in patients with rheumatoid arthritis in 1993 [49]. Its use in patients with IBD has revolutionized the treatment of those patients by being efficient in inducing remission and sparing steroids.
Infliximab is bivalent IgG monoclonal antibody. It is a chimeric antibody and contains 25% mouse-derived amino acids in the binding regions and 75% human-derived amino acids in the constant regions of the molecule. Adalimumab and golimumab are fully humanized bivalent IgG monoclonal antibodies. Certolizumab pegol is different as it is not a complete antibody but consists in a monovalent Fab antibody fragment covalently linked to polyethylene glycol. In contrast to the other three substances, certolizumab does not activate complement because as a Fab fragment it does not contain an Fc region [4].
There are also pharmacokinetic differences that play a role in the immunogenicity of these substances. Infliximab is given intravenously every 8 weeks. Its administration leads to higher initial concentrations and lower trough levels in contrast to the more frequently applied substances adalimumab, golimumab and certolizumab pegol [50].
How do TNF antagonists work?
Although commonly used in the clinic for the treatment of IBD, the precise mechanism of action of TNF antagonists is not known (Figure 5). TNF antagonists may (i) block the mTNF-TNFR1/2- interaction (acting as an antagonist), (ii) induce reverse signaling (acting as an agonist), (iii) induce cell cytotoxicity by activating antibody-dependent cell-mediated cytotoxicity, or by activating complement-dependent cytotoxicity through their Fc-region, (iv) inactivates TNF. mTNF-TNFR1/2-interaction induces the production of inflammatory cytokines but also induces apoptosis / necroptosis depending on the tissue context. Blocking mTNF-TNFR1/2- interactions by TNF antagonists leads to reduced expression of proinflammatory cytokines, such as IL-6 [51]. TNF antagonists induced reverse signaling leads to apoptosis, which may contribute to the deletion of macrophages producing pro-inflammatory cytokines. Binding to Fc receptors mediates antibody-dependent cell-mediated cytotoxicity. This leads to the clearance of immune cells, which play an essential role for the development and persistence of inflammation. All TNF antagonists inactivate sTNF. But this mechanism seems not to play a major role in IBD, as Etanercept is ineffective for the treatment of Crohn’s disease. This means that the role of TNF antagonists for the treatment is likely mediated by blocking mTNFTNFR1/ 2-interaction, induction of reverse signaling and Fc- (cellor complement-) dependent cytotoxicity. This participates in the restoration of the integrity of the tight junctions of the intestinal epithelium, down-regulation of mucosal angiogenesis, induction of apoptosis of T cells prevention of chemotaxis of inflammatory cells and recruitment of immune-suppressive cells to the inflamed intestine [52].

Figure 5: Mechanisms of action of TNF antagonists.
When are TNF antagonists used?
Although TNF antagonists are used in a wide variety of autoimmune and inflammatory disorders we will focus on patients with IBD. In patients with an IBD, they are used in moderate to severe disease activity, in steroid dependent or steroid refractory disease course, fistulizing and extensive Crohn’s disease. There is still a debate when TNF antagonists should be used for the treatment of Crohn’s disease and ulcerative colitis. Early use may influence the disease course, but on the other side overtreatment of patients with mild symptoms should be avoided.
All currently available TNF antagonists seem to have a similar efficacy. Here the results of the most important studies are shown, but the response rates and the remission rates cannot be compared directly due to different inclusion criteria and study design (Table 2).
![]()
Crohn’s disease
Definition of clinical response
Response rate
Definition of remission
Remission rate
Infliximab
[84]
Reduction of CDAI of 70 points or more
Response rate: 81% after a single infusion
Clinical remission defined as a CDAI<150
Remission rate: 24% (not significant)
[85]
Decrease of the CDAI >70 at week 2, week 30 and week 54
Response rate: 62%
Remission defined as a CDAI <150 and discontinuation of steroids at week 2, week 30 and week 54
Remission rate:
39% (5 mg/kg) - 45% (10 mg/kg)
Adalimumab
[86]
Decrease of the CDAI >70, at week 4
Response rate: 50%
Defined as a CDAI <150, at week 4
Remission rate: 36%
Certolizumabpegol
[87]
Decrease of the CDAI >70, up to week 26
Response rate: 35%
CDAI <150, up to week 26
Remission rate: same as placebo
Ulcerative Colitis
Definition of clinical response
Response rate
Definition of remission
Remission rate
Infliximab
[88]
Decrease of the Mayo score of at least 3 points, week 8, 30 and 54
Response rate: 61-69%
Mayo score of 2 and lower, week 8, 30 and 54
Remission rate: 27.5 - 38.8%
Adalimumab
[89]
Decrease of the Mayo score of at least 3 points, week 8, 32 and 52
Response rate: 50.4%
Mayo score of 2 and lower, week 8, 32 and 52
Remission rate: 16.5%
Golimumab
[90]
Decrease of the Mayo score of at least 3 points, week 6
Response rate 54.9%
Mayo score of 2 and lower, week 6
Remission rate 18.7%
Table 2: Response and remission rates of the available TNF antagonists.
What side effects have been described for TNF antagonists used?
Patients treated with TNF antagonists have an increased risk for the development of infections, infusion reactions, hematological adverse events, neurological adverse events and autoimmune adverse events (likely caused by paradoxical inflammation). A recently published systematic review about the safety of anti-TNF treatment in patients with rheumatoid arthritis showed that infections, particularly reactivation of latent tuberculosis or de novo tuberculosis, infusion reactions and abnormalities in hepatic liver enzymes are the most common side effects in patients treated with infliximab [53]. The main adverse effects of adalimumab were infections and infusion reactions. Other side effects include neurological adverse events (mainly demyelinating diseases) and hematological adverse events such as cytopenia. The relationship between TNF antagonists and cancer remains controversial despite many comprehensive reports. Williams, et al. [54] conducted a systematic review in which all available placebo-controlled studies that compared anti-TNF therapy with placebo in adults with Crohn’s disease and ulcerative colitis were included, involving 7054 patients. They did not find an increased risk of malignancy in patients under anti-TNF therapy compared to patients receiving placebo. Another study investigated the risk in patients with Crohn’s disease receiving adalimumab and also found that patients receiving adalimumab mono therapy did not have a greater than expected risk of cancer (non-melanoma skin cancer and other cancers) [55]. In contrast, this study found that the patients receiving a combination therapy had a 5-fold risk of non-melanoma skin cancers and a 3-fold risk of other cancers and even a 8-fold risk of lymphoma compared to the general population. This increased risk was attributed to the immunomodulator. Autoimmune adverse events are common events. The lupus-like syndrome was described to evolve in up to 1.75% in patients with spondyloarthritis treated with TNF antagonists. Psoriasis is another common autoimmune adverse event of the TNF antagonists. Rarely, other side effects such as interstitial lung disease and sarcoidosis have been described.
What is paradoxical inflammation induced by TNF antagonists?
TNF antagonists are successfully used to treat diseases with an immunological background. Besides the more frequent side effects as allergies and infections, the new onset of other immunological diseases can occur such as psoriasis, arthritis, and lupus-like reactions. Even new onset IBD has been observed in patients with arthritis treated with TNF antagonists [17]. This phenomenon is termed “paradoxical inflammation”. The exact pathophysiological mechanism of TNF induced inflammation is not known. Furthermore, whether the pathophysiology of paradoxical inflammation differs between IBD and non-IBD patients is not known. Besides binding with their Fab (antigen-binding region) fragment to TNF, infliximab and adalimumab also bind with their Fc (crystallizable fragment) region to the Fc receptors CD64 (Fc-gamma receptor I (FcγRI)) and CD16/32 (Fc-gamma receptor III/II (FcγRIII/II)) expressed by monocytes and macrophages. This may induce Son of Sevenless (SOS) and p38 activation, which in turn will lead to the secretion of IL-12/IL-23 [56]. Switching of infliximab to certolizumab dose not fully resolve paradoxical inflammation. The activation of intracellular signaling cascades via Fc receptors may hence not fully explain the development of paradoxical inflammation in patients treated with TNF antagonists. Below we will discuss the most frequently observed entities and the underlying current pathophysiologic concepts.
Psoriasis: Despite the fact that TNF antagonists are potent drugs in treatment-refractory psoriasis, psoriatic skin lesions have been observed as a class effect of these drugs [57]. A recent publication from Tillack, et al. [58] has revealed new insights in the pathomechanisms of this paradoxical phenomenon.
Epidemiology: This side effect occurred in 4.8% of 434 patients of this prospective study. The authors found that smoking, increased body mass index and a short IBD duration were the main predictors. The clinical picture was different to that observed in patients with genuine psoriasis (psoriasis in patients without anti-TNF treatment). The most common form was palmoplantar lesions (11/21 patients) and guttate psoriasis (7/21 patients). 9/21 patients had scalp lesions; three of these patients developed a diffuse alopecia. In other case series the Massachusetts General Hospital in Boston [59] the occurrence of psoriasis was more frequently described in women than in men.
Pathomechanism: The pathogenesis has been largely unknown. Seneschal, et al. [60] examined 13 patients with TNF antagonistinduced psoriasiform lesions. According to their results, they hypothesized that psoriasiform lesions triggered by TNF antagonists are different from true psoriasis. They found strong production of MxA, a specific marker for the production of type I IFN, in inflammatory cells, a cytokine for which beneficial effects in colitis models has been described. The type I interferon (IFN) IFN-β suppress Th1 mediated experimental autoimmune encephalomyelitis (EAE) and multiple sclerosis but augments Th17- mediated EAE. The expression of MxA was associated with an increased expression of CXCL9, which plays a role in the migration of CXCR3+ lymphocytes into the skin. IFN-a plays a major role in psoriasis. Possibly blocking TNF-a increases the production of IFN-a by plasmacytoid dendritic cells by inhibiting the negative feedback of TNF on pDC generation and maturation. Tillack, et al. showed in their study that all patients with severe lesions in their cohort were G/G wild type carriers for the rare coding IL23R variant rs11209026. This variant is associated with increased Th17 cytokine production. Their immunohistochemical analysis showed infiltrates of IL-17A-secreting T cells and IL-22-secreting T-cells in the affected skin. This cytokine pattern is characteristic of Th17 cells. Infiltrates of IFN-γ-expressing Th1 cells were found in all patients indicating that these Th1 and Th17 cells may play a major role in the pathogenesis of TNF antagonist-induced psoriasiform skin lesions.
Treatment: The first-line treatment is a topical treatment, consisting mainly of topical glucocorticoids, but this is often not efficacious. 33.3% of patients seems not improve under topical therapy. There are several possibilities in patients not improving under topical treatment. The switch to a second and third TNF antagonist is usually not effective. In a retrospective 19/62 patients with psoriatic skin lesions induced by TNF antagonists were switched to another TNF antagonist with improvement in one patient only [61]. Two patients were also switched unsuccessfully to a third TNF antagonist. Overall recurrent and severe psoriatic skin lesions lead to 40% discontinuation of TNF-antagonists despite the concomitant treatment with topical corticosteroids, keratolytics, emollients, vitamin D analogues and ultraviolet (UV) therapy. The rash reappeared after the switch to another TNF antagonist in 52% of the patients. 23% of the patients had eventually to stop the treatment with TNF antagonists. The addition of methotrexate in patients with TNF antagonist-induced psoriasiform lesions has no benefit [62]. TNF antagonists have to be stopped in the patients who are refractory to topical treatment, and these patient scan be treated with Ustekinumab, an anti-IL12/IL-23 antibody. IL-12 is of importance for the development of Th1 cells. TGF-β and IL-6 are required for the differentiation of TH17 cells, which then express surface IL-23R. IL-23 then stabilizes and expands Th17 cells. Ustekinumab interferes with Th1 and Th17 driven disease. All 7 patients switched to Ustekinumab showed a marked improvement of their skin lesions. Remission of CD was maintained in 5/6 patients and 2/3 patients with active disease had a significant clinical improvement, which correlates with the clinical efficacy of this drug in CD patients [63]. Here as well, switching to another TNF antagonist was not a good option.
Alopecia: Numerous case reports and case series have described the development of Alopecia areata in patients treated with anti- TNF agents. The biggest report based on a French Pharmaco vigilance database was 52 (4.9%) of 1065 alopecia after Anti-TNF agents (18 involved infliximab, 17 adalimumab, 15 etanercept and 2 certolizumab). The reporting odd ratio for the development of alopecia during exposure to TNF-a antagonists was 3.0, (95% CI, 2.3 - 4.0). This dermatological complication remains however quite rare with 18 additional clinical cases reported in the literature according to a review in 2012 [64]. The hair loss occurred mostly in the scalp but could extend to the eyebrow, axilla and pubis.
Pathomechanism: The pathogenesis is linked to the induction or aggravation of an autoimmune reaction.
Treatment: The first-line treatment is mainly to stop the drug, which reversed the process in most cases. However, when in some cases alopecia persists, this could be difficult to accept in particular by female patients.
Eczema: Eczematiform reactions belong to the allergic reactions that occur under anti-TNF agents. Moreover it seems to occur to patients with a personal and/or family history of atopy.
Epidemiology: Eczema has been reported in up to 7% of rheumatoid arthritis patients receiving biological agents against TNF. Rahier, et al. described 23 eczematiform lesions in their review of 85 IBD patients and in the same paper estimate the prevalence based in the Lille cohort at 3% [61].
Pathomechanism: Is thought to be similar to the one described of psoriasis.
Treatment: Most patients seem to respond adequately to topical therapy (steroids, emollient and in some cases tacrolimus). When the patients do not respond to topical therapies, then it should be considered if the anti-TNF agent has to be stopped.
Arthritis: Arthralgia is a frequent complaint in IBD patients. Usually they occur in the context of IBD-associated arthropathy. Less commonly, the treatment with TNF antagonists induces arthritis as a paradoxical side effect.
Epidemiology: Among the few reports about TNF-antagonistinduced paradoxical joint inflammation, one case series, published as an abstract, suggested an incidence of 1.6% in IBD patients receiving infliximab or adalimumab to develop arthritis [65]. In some but not all patients a high ANA titer and an elevated anti-ds-DNA titer was observed, so that it is difficult to distinguish paradoxical arthritis from lupus-like reaction [57]. Clinical criteria, such as serositis, involvement of kidneys, skin rash, have to be in addition considered to distinguish arthritis from lupus-like reactions. Interestingly, there was no correlation with antibodies to infliximab. New onset arthritis as a paradoxical side effect of treatment with TNF antagonists is better known in patients with hidradenitis suppurativa, another chronic inflammatory disease, treated with TNF antagonists in severe treatment-refractory cases. Three out of 11 patients (27%) with severe HS treated with infliximab developed an acute and painful polyarthritis without systemic reaction, leading to the discontinuation of infliximab therapy as reported in a retrospective study [66].
Pathomechanism: The pathomechanism is not known. It has been hypothesized that the blockade of TNF leads to a disturbed balance of inflammatory and regulatory cytokines and cells. Delayed hypersensitive reactions have also been proposed.
Treatment: Up to now, the therapeutic strategy for patients with paradoxical arthritis has not been well established. In the Leuven case series neither the dose escalation of the TNF antagonist nor the switch to another TNF antagonist improved the symptoms. The discontinuation of the TNF antagonist, the initiation of corticosteroids and the addition of an immunomodulator improved the symptoms.
Lupus like reactions: Treatment with TNF antagonists leads to the formation of antinuclear antibodies (ANA) in a high number of patients (increase from 24% to 77% after 30 weeks in patients with rheumatoid arthritis in the prospective study by Eriksson, et al. [67], 56.8% in patients with Crohn’s disease in the study by Vermeire, et al. [16]. However, only a minority of the patients with ANA develops the clinical picture of drug-induced lupus erythematosus (DILE). The incidence of DILE in patients treated with TNF antagonists seems to range from 0.19% up to 1.6% [16,68].
Pathomechanism: Several mechanisms have been proposed how these autoantibodies are induced. Possibly, the induction of apoptosis or necroptosis by TNF antagonists leads to the release of cell products. The released cell material, especially nucleosomes, can immunize the host, which may result in the production of antinuclear antibodies. Deregulation of apoptosis or inhibition of a cytotoxic T-lymphocyte response that normally suppresses auto reactive B-cells also seems to play a role [69].
The clinical picture encompasses mainly joint manifestations, especially symmetric large joint arthralgia, synovitis, fever, rashes, leukopenia and rarely renal involvement. The antinuclear titer is frequently elevated. Typically anti-dsDNA and/or anti-nucleosome antibodies are tested positive. Anti-nucleosome antibodies of the IgG class even seem to be a more sensitive marker of SLE than anti-ds- DNA antibodies.
There are no clearly established diagnostic criteria for the diagnosis of drug-induced lupus erythematosus. A French group suggested that as least 4 ACR criteria with a clear timely relationship with the TNF antagonist treatment is required for the diagnosis of drug-induced lupus erythematosum, which is supported by high ANA titers. After discontinuation of the treatment with TNF antagonists the symptoms have to disappear. An additional feature of drug-induced lupus erythematosus is the presence of low serum complement [68].
Treatment: The treatment of the drug-induced lupus like syndrome consists in stopping the inducing agent. In a cohort of 13 patients with infliximab-induced DILE joint manifestations dominated, in which the re-challenge with a further TNF antagonist is associated with a low rate of recurrence [70].
Vasculitis: Vasculitis is a rare but potentially fatal side effect of TNF antagonists. Review of the current literature identified 113 patients with a vasculitis as a side effect of the therapy with TNF antagonists [71]. The main manifestation was cutaneous in 87%, including purpura, ulcerative lesions, nodules, digital vasculitis, and macro papular rash. As many as one quarter of the patients had extra cutaneous manifestations including visceral vasculitis (24%), peripheral nerve involvement (16%), renal involvement (13%), central nervous involvement (4%), and lung involvement (3%). In 24% the ANA were elevated, in 9% the ANCA were elevated and also elevated cryoglobulins, anti-DNA, anti-phospholipid antibodies and anti-Ro/La were reported. A more recently published report from the Mayo Clinic included also many patients with IBD. This retrospective review identified 8 patients with a TNF antagonistassociated vasculitis. In these patients, the skin was involved in 63%, the peripheral nerve in 25%, and the kidney in 1 patient (13%) [72]. As already mentioned, TNF antagonist-associated vasculitis is rare. Because it can be fatal, healthcare professionals should nonetheless be aware of this entity and, in the case of suspect, try to make a diagnosis by histological examination and conduct extensive investigations for organ involvement.
Pathomechanism: Several hypotheses are discussed. The deposition of immunocomplexes consisting of the drug and its target could induce the local activation of the complement system. Another hypothesis is the occurrence of a cytokine imbalance in patients treated with TNF antagonists.
Treatment: The treatment consists in stopping the TNF antagonist. In a retrospective study the vasculitis improved in 63% of patients after stopping the TNF antagonist [73]. Patients not improving after stopping the offending drug have been successfully treated with high-dose corticosteroids and/or immunosuppressant drugs, such as cyclophosphamide, mycophenolate mofetil, hydroxychloroquine, methotrexate, azathioprine or rituximab [72,73].
Sarcoidosis: TNF plays a key role in the formation of granulomas, and TNF antagonists have been showed to be an efficient treatment in treatment-refractory sarcoidosis [74]. Despite these facts, sarcoidosis can also occur as a paradoxical side effect by TNF antagonists, mainly in patients with arthritis. In a call for observations of sarcoid-like granulomatosis following anti-TNF therapy along the members of the French “Club Rhumatismeset Inflammation” and were able to identify 10 patients with this side effect [75]. They calculated a frequency of at least 0.04%. The patients in this report suffered from pulmonary (cough, dyspnea) and cutaneous symptoms (cutaneous nodules, erythema nodosum, hypodermitis and scar inflammation). Three of the patients also had general symptoms such as weight loss, fatigue and fever. The review of the Australian Biologics clinic database showed an ever-higher frequency of new-onset sarcoidosis under TNF antagonists of 1.82%, though it has to be taken into account that in these patients, methotrexate was continued and its use could have a contributory role in these cases [76]. There are also case reports in patients with IBD showing the development of sarcoidosis in these patients [77], but it has to be pointed out that most cases were observed in patients using etanercept and that these observations are hence less applicable to IBD patients [78].
Pathomechanism: The development of granulomas in patients treated with TNF antagonists is paradoxical, and its etiology is unclear. It has been hypothesized that a modulation of the cytokine network by TNF antagonists may restore a Th1 response and restore IFN-γ, another key player in granuloma formation. Another hypothesis is that the increased susceptibility to infection under TNF antagonists triggers granuloma formation, as the development of sarcoidosis requires the exposure to a triggering antigen.
Diagnostics: Sarcoidosis must be considered when a patient under TNF antagonist treatment develops cough or dyspnea, when an infection, most importantly tuberculosis, has been ruled out. In the lab, hypercalcemia and an increased ACE activity can be found. In pulmonary sarcoidosis, the most frequent organ manifestation, the chest CT scan shows mediastinal and/or hilar adenopathies and lung infiltrates. In cutaneous sarcoidosis, skin biopsy leads to the diagnosis.
Treatment: Stopping of the TNF antagonists with or without administration of systemic corticosteroids may lead to clinical improvement in all of the patients [75].
De novo inflammatory bowel diseases: It seems always difficult to assess exacerbation of previously diagnosed IBD associated with anti- TNF inhibition. However multiple registers based on rheumatological diseases help to identify this phenomenon. As probably each IBD specialized university center has seen one or two highly suggestive cases, a nice example of drug re-challenge with etanercept have been published. This manuscript reported a case with juvenile rheumatoid arthritis, who developed Crohn’s -like disease under treatment with etanercept, which resolved after TNF antagonists were stopped [79]. Distinguishing anti-TNF induced IBD from ongoing IBD in a given patient may be difficult. The patient’s history needs to be considered to distinguish anti-TNF induced IBD from ongoing IBD.
Epidemiology: These cases represent mostly Crohn’s or Crohn’s like diseases or unspecified colitis. Patients with juvenile arthropathies are more likely to development of de novo IBD whereas the culprit drug is more frequently etanercept [80] an all-human soluble TNF-receptor that is fused to the Fc portion of IgG1 or infliximab/ adalimumab. In a study using the food and drug administration adverse events Reporting System [81], 158 cases were identified and the association was considered for 111 “possible” and 47 “ “probable” using the Navajo algorithm [82]. However, the natural history of rheumatological disease is also associated with gut inflammation (e.g. being subclinical in about 40-60% of patients with ankylosing spondylitis) as extra-articular manifestations of the disease, which end up to a definitive IBD diagnosis in about 10% of them [83].
Pathomechanism: Paradox inflammation induced by TNF inhibition, given that neutralization of TNF-a or absence of TNF-a- exacerbated acute DSS-induced colitis in animal models.
Therapy: Ceasing the offending drug and treating the inflammatory bowel disease when possible with alternative options.
Hidradenitis suppurativa: Hidradenitis suppurativa (HS) is a painful chronic relapsing inflammatory disease of the skin characterized by inflammatory nodules and abscesses, which can lead to scares of areas bearing the apocrine glands. HS can be associated with IBD, mainly Crohn’s disease, and is treated with anti-TNF agents, such as adalimumab. Paradoxically, HS can also occur in patients treated with anti-TNF agents. After stopping the anti-TNF agents or switch to another biological HS resolves.
Epidemiology: A French retrospective multicenter study from May 2004 to July 2015 has reported 25 patients, who developed HS under treatment with anti-TNF agents for rheumatoid arthritis (7 patients), Crohn’s disease (9 patients, chronic juvenile arthritis (1 patient), ankylosing spondylitis (5 patients), SAPHO syndrome (1 patient), psoriasis (1 patient) and psoriasis arthritis (1 patient).
Pathomechanism: The pathophysiology of HS induced by anti- TNF agents has not been resolved.
Therapy: Stopping the anti-TNF agents or switching to other biological is required to resolve HS.
Rare autoimmune diseases: Rarely the development of interstitial lung disease (interstitial pneumonitis, pulmonary hemorrhage, and bronchiolitis obliterans organizing pneumonia), inflammatory myopathies and antiphospholipid syndrome after treatment with anti-TNF therapy has been described [71].
Conclusion
Paradoxical inflammation induced by TNF antagonists is a side effect more common than previously suspected. Their pathogenesis is widely unknown, but an imbalance of the pro-inflammatory and regulative cytokines, induced by a blockade of TNF, seems to play an important role. In particular type I IFNs and IL-17A producing Th17 cells are essential for the development of paradoxical inflammation.
In the daily clinical practice, the most common encountered phenomenon is a rise in the titer of autoimmune antibodies, especially the ANA titer. In most cases elevated ANA titer is not associated with clinical symptoms. Less frequently the immunogenicity of the TNF antagonists leads to diseases as drug-induced lupus, psoriasis and vasculitis. Despite the fact that they are rare, their recognition is important as they can potentially be fatal and need appropriate treatment.
Depending on the clinical entity and severity there are different treatment options. Stopping the culprit agent is the causative therapy. The underlying disease requires efficient treatment. The TNF antagonist has to be substituted by another TNF antagonist, especially certolizumab, which is less immunogenic. However, the switch of a TNF antagonist to certolizumab (lacking the Fc regions) did not improve TNF induced psoriasis in IBD patients. This means, that the occurrence of paradoxical inflammation is a class effect of TNF antagonists [58]. In some circumstances it is possible to add an immunosuppressant drug and continue with the TNF antagonist, e.g. in arthritis. Likely, the TNF antagonist has to be replaced by an agent of a different class, e.g. the recently available integrin inhibitor vedolizumab. Up to now there are no immunological reactions to this drug, albeit its efficiency in patients developing anti-TNF-induced immunological reactions has not been evaluated yet. The IL 12/23 antagonist Ustekinumab has been shown to be efficient in patients with treatment-refractory psoriasis and Crohn’s disease and is especially useful in patients with psoriasiform side effects.
References
- Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004; 15(5): 353-66.
- Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012; 119: 651-65.
- Vilcek J. From IFN to TNF: a journey into realms of lore. Nat Immunol. 2009; 10: 555-557.
- Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008; 117: 244-279.
- Kang YM, Zhang ZH, Johnson RF, Yu Y, Beltz T, Johnson AK, et al. Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res. 2006; 99: 758-766.
- Argilés JM, López-Soriano FJ. The role of cytokines in cancer cachexia. Med Res Rev. 1999; 19: 223-248.
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001; 344: 907-916.
- Feldmann M, Maini RN. Anti-TNF therapy, from rationale to standard of care: what lessons has it taught us? J Immunol. 2010; 185: 791-794.
- Winston BW, Krein PM, Mowat C, Huang Y. Cytokine-induced macrophage differentiation: a tale of 2 genes. Clin Invest Med. 1999; 22: 236-255.
- Webb NJ, Myers CR, Watson CJ, Bottomley MJ, Brenchley PE. Activated human neutrophils express vascular endothelial growth factor (VEGF). Cytokine. 1998; 10: 254-257.
- Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord. 2003; 27: S53-S55.
- Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A. Tumor necrosis factor alpha-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J Biol Chem. 1995; 270: 23780-23784.
- Shrivastava AK, Singh HV, Raizada A, Singh SK, Pandey A, Singh N, et al. Inflammatory markers in patients with rheumatoid arthritis. Allergol Immunopathol (Madr). 2015; 43: 81-87.
- Murch SH, Lamkin VA, Savage MO, Walker-Smith JA, MacDonald TT. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut. 1991; 32: 913-917.
- Fiorino G, Danese S, Pariente B, Allez M. Paradoxical immune-mediated inflammation in inflammatory bowel disease patients receiving anti-TNF-alpha agents. Autoimmun Rev. 2014; 13: 15-19.
- Vermeire S, Noman M, Van Assche G, Baert F, Van Steen K, Esters N, et al. Autoimmunity associated with anti-tumor necrosis factor alpha treatment in Crohn's disease: a prospective cohort study. Gastroenterology. 2003; 125: 32-39.
- Fouache D, Goeb V, Massy-Guillemant N, Avenel G, Bacquet-Deschryver H, Kozyreff-Meurice M, et al. Paradoxical adverse events of anti-tumour necrosis factor therapy for spondyloarthropathies: a retrospective study. Rheumatology (Oxford). 2009; 48: 761-764.
- Aubin F, Carbonnel F, Wendling D. The complexity of adverse side-effects to biological agents. J Crohns Colitis. 2013; 7: 257-262.
- Pichler WJ. Adverse side-effects to biological agents. Allergy. 2006; 61: 912-920.
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975; 72: 3666-3670.
- Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015; 15: 362-374.
- Noti M, Corazza N, Mueller C, Berger B, Brunner T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med. 2010; 207: 1057-1066.
- Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol. 2006; 176: 721-725.
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009; 16: 3-11.
- Tartaglia LA, Ayres TM, Wong GH, Goeddel DV. A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993; 74: 845-853.
- Lavrik I, Golks A, Krammer PH. Death receptor signaling. J Cell Sci. 2005; 118: 265-267.
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995; 81: 495-504.
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996; 4: 387-396.
- Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996; 84: 299-308.
- Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006; 281: 13636-13643.
- Sasaki K, Iwai K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol Rev. 2015; 266: 175-189.
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NF-kappa B activation. Nat Cell Biol. 2009; 11: 123-132.
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001; 412: 346-351.
- Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004; 15: 535-548.
- Israël A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010; 2: a000158.
- Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995; 269: 1424-1427.
- Enesa K, Zakkar M, Chaudhury H, Luong le A, Rawlinson L, Mason JC, et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem. 2008; 283: 7036-45.
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004; 430: 694-699.
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009; 137: 1100-1111.
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008; 133: 693-703.
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010; 11: 700-714.
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012; 150: 339-350.
- Watts AD, Hunt NH, Wanigasekara Y, Bloomfield G, Wallach D, Roufogalis BD, et al. A casein kinase I motif present in the cytoplasmic domain of members of the tumour necrosis factor ligand family is implicated in 'reverse signalling'. EMBO J. 1999; 18: 2119-2126.
- Eissner G, Kirchner S, Lindner H, Kolch W, Janosch P, Grell M, et al. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol. 2000; 164: 6193-6198.
- Atreya R, Neumann H, Neufert C, Waldner MJ, Billmeier U, Zopf Y, et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn's disease. Nat Med. 2014; 20: 313-318.
- Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin Exp Immunol. 1993; 94: 174-181.
- Slebioda TJ, Kmiec Z. Tumour necrosis factor superfamily members in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 2014; 2014: 325129.
- Kaser A. Not all monoclonals are created equal - lessons from failed drug trials in Crohn's disease. Best Pract Res Clin Gastroenterol. 2014; 28: 437-449.
- Elliott MJ, Maini RN, Feldmann M, Long-Fox A, Charles P, Katsikis P, et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993; 36: 1681-1690.
- Jinesh S. Pharmaceutical aspects of anti-inflammatory TNF-blocking drugs. Inflammopharmacology. 2015; 23: 71-77.
- Ringheanu M, Daum F, Markowitz J, Levine J, Katz S, Lin X, et al. Effects of infliximab on apoptosis and reverse signaling of monocytes from healthy individuals and patients with Crohn's disease. Inflamm Bowel Dis. 2004; 10: 801-810.
- Peake ST, Bernardo D, Mann ER, Al-Hassi HO, Knight SC, Hart AL. Mechanisms of action of anti-tumor necrosis factor alpha agents in Crohn's disease. Inflamm Bowel Dis. 2013; 19: 1546-1555.
- Nanau RM, Neuman MG. Safety of anti-tumor necrosis factor therapies in arthritis patients. J Pharm Pharm Sci. 2014; 17: 324-361.
- Williams CJ, Peyrin-Biroulet L, Ford AC. Systematic review with meta-analysis: malignancies with anti-tumour necrosis factor-alpha therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2014; 39: 447-458.
- Osterman MT, Sandborn WJ, Colombel JF, Robinson AM, Lau W, Huang B, et al. Increased risk of malignancy with adalimumab combination therapy, compared with monotherapy, for Crohn's disease. Gastroenterology. 2014; 146: 941-949.
- Niess JH, Danese S. Anti-TNF and skin inflammation in IBD: a new paradox in gastroenterology? Gut. 2014; 63: 533-535.
- Cleynen I, Vermeire S. Paradoxical inflammation induced by anti-TNF agents in patients with IBD. Nat Rev Gastroenterol Hepatol. 2012; 9: 496-503.
- Tillack C, Ehmann LM, Friedrich M, Laubender RP, Papay P, Vogelsang H, et al. Anti-TNF antibody-induced psoriasiform skin lesions in patients with inflammatory bowel disease are characterised by interferon-γ-expressing Th1 cells and IL-17A/IL-22-expressing Th17 cells and respond to anti-IL-12/IL-23 antibody treatment. Gut. 2014; 63: 567-577.
- Cullen G, Kroshinsky D, Cheifetz AS, Korzenik JR. Psoriasis associated with anti-tumour necrosis factor therapy in inflammatory bowel disease: a new series and a review of 120 cases from the literature. Aliment Pharmacol Ther. 2011; 34: 1318-1327.
- Seneschal J, Milpied B, Vergier B, Lepreux S, Schaeverbeke T, Taïeb A. Cytokine imbalance with increased production of interferon-alpha in psoriasiform eruptions associated with antitumour necrosis factor-alpha treatments. Br J Dermatol. 2009; 161: 1081-1088.
- Rahier JF, Buche S, Peyrin-Biroulet L, Bouhnik Y, Duclos B, Louis E, et al. Severe skin lesions cause patients with inflammatory bowel disease to discontinue anti-tumor necrosis factor therapy. Clin Gastroenterol Hepatol. 2010; 8: 1048-1055.
- Buisson A, Cuny JF, Barbaud A, Schmutz JL, Bigard MA, Gueant JL, et al. Methotrexate for psoriasiform lesions associated with anti-tumour necrosis factor therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2012; 35: 1175-1180.
- Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. 2012; 367: 1519-1528.
- Navarro R, Dauden E, Gallo E, Santiago Sanchez-Mateos D, Garcia-Diez A. Alopecia areata during treatment of psoriasis with adalimumab and leflunomide: a case and review of the literature. Skin Pharmacol Physiol. 2012; 25: 107-110.
- Van Moerkercke W, Ackaert C, Jürgens M, Kasran A, Compernolle G, Ballet V, et al. 407 Anti-TNFa Induced Severe Arthralgia as a Manifestation of Autoimmunity?. Gastroenterology. 2010; 138: 60-61.
- Acquacalda E, Roux CH, Albert C, Breuil V, Passeron T, Euller-Ziegler L. New onset of articular inflammatory manifestations in patients with hidradenitis suppurativa under treatment with infliximab. Joint Bone Spine. 2015; 82: 362-364.
- Eriksson C, Engstrand S, Sundqvist KG, Rantapää-Dahlqvist S. Autoantibody formation in patients with rheumatoid arthritis treated with anti-TNF alpha. Ann Rheum Dis. 2005; 64: 403-407.
- De Bandt M, Sibilia J, Le Loet X, Prouzeau S, Fautrel B, Marcelli C, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005; 7: R545-R551.
- D'Auria F, Rovere-Querini P, Giazzon M, Ajello P, Baldissera E, Manfredi AA, et al. Accumulation of plasma nucleosomes upon treatment with anti-tumour necrosis factor-alpha antibodies. J Intern Med. 2004; 255: 409-418.
- Subramanian S, Yajnik V, Sands BE, Cullen G, Korzenik JR. Characterization of patients with infliximab-induced lupus erythematosus and outcomes after retreatment with a second anti-TNF agent. Inflamm Bowel Dis. 2011; 17: 99-104.
- Ramos-Casals M, Brito-Zeron P, Munoz S, Soria N, Galiana D, Bertolaccini L, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007; 86: 242-251.
- Sokumbi O, Wetter DA, Makol A, Warrington KJ. Vasculitis associated with tumor necrosis factor-a inhibitors. Mayo Clin Proc. 2012; 87: 739-745.
- Saint Marcoux B, De Bandt M; CRI (Club Rhumatismes et Inflammation). Vasculitides induced by TNFalpha antagonists: a study in 39 patients in France. Joint Bone Spine. 2006; 73: 710-713.
- Ziegenhagen MW, Müller-Quernheim J. The cytokine network in sarcoidosis and its clinical relevance. J Intern Med. 2003; 253: 18-30.
- Daien CI, Monnier A, Claudepierre P, Constantin A, Eschard JP, Houvenagel E, et al. Sarcoid-like granulomatosis in patients treated with tumor necrosis factor blockers: 10 cases. Rheumatology (Oxford). 2009; 48: 883-886.
- Tong D, Manolios N, Howe G, Spencer D. New onset sarcoid-like granulomatosis developing during anti-TNF therapy: an under-recognised complication. Intern Med J. 2012; 42: 89-94.
- Takahashi H, Kaneta K, Honma M, Ishida-Yamamoto A, Ashida T, Kohgo Y, et al. Sarcoidosis during infliximab therapy for Crohn's disease. J Dermatol. 2010; 37: 471-474.
- Toussirot E, Aubin F. Paradoxical reactions under TNF-alpha blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016; 2: e000239.
- Zeitz J, Enderlin S, Biedermann L, Turina M, Leibl S, Prakash M, et al. New Onset, Aggravation and Recurrence of Crohn's Disease upon Treatment with Three Different Tumor Necrosis Factor Inhibitors. Case Rep Gastroenterol. 2015; 9: 106-112.
- van Dijken TD, Vastert SJ, Gerloni VM, Pontikaki I, Linnemann K, Girschick H, et al. Development of inflammatory bowel disease in patients with juvenile idiopathic arthritis treated with etanercept. J Rheumatol. 2011; 38: 1441-1446.
- Krishnan A, Stobaugh DJ, Deepak P. Assessing the likelihood of new-onset inflammatory bowel disease following tumor necrosis factor-alpha inhibitor therapy for rheumatoid arthritis and juvenile rheumatoid arthritis. Rheumatol Int. 2015; 35: 661-668.
- Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981; 30: 239-245.
- Rudwaleit M, Baeten D. Ankylosing spondylitis and bowel disease. Best Pract Res Clin Rheumatol. 2006; 20: 451-471.
- Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997; 337: 1029-1035.
- Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002; 359: 1541-1549.
- Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006; 130: 323-333.
- Sandborn WJ, Feagan BG, Stoinov S, Honiball PJ, Rutgeerts P, Mason D, et al. Certolizumab pegol for the treatment of Crohn's disease. N Engl J Med. 2007; 357: 228-238.
- Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005; 353: 2462-2476.
- Sandborn WJ, van Assche G, Reinisch W, Colombel JF, D'Haens G, Wolf DC, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012; 142: 257-265.
- Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014; 146: 85-95.