
Research Article
Austin J Gastroenterol. 2019; 6(2): 1102.
25(OH)-D3 Alleviate Liver NK Cytotoxicity in Acute but not in Chronic Fibrosis Model of BALB/C Mice due to Modulations in Vitamin D Receptor
Salhab A, Amer J* and Safadi R
Research Center for Liver Diseases, Liver & Gastroenterology Units; Division of Medicine, Hadassah– Hebrew University Hospital- Jerusalem, Israel
*Corresponding author: Johnny Amer, Research Center for Liver Diseases, Liver & Gastroenterology Units; Division of Medicine, Hadassah–Hebrew University Hospital- Jerusalem, Israel
Received: July 10, 2019; Accepted: August 27, 2019; Published: September 03, 2019
Abstract
Background: Low 25-Hydroxy-vitamin-D; “25(OH)-D3” serum and vitamin D receptor (VDR) levels were recently correlated to advanced fibrosis. However, VDR mechanism in liver fibrosis modulations is not well understood.
Aim: To evaluate associations of VDR with NK cell modulations during fibrogenesis.
Methods: Carbon-tetrachloride (CCl4) hepatic-fibrosis was induced in BALB/c mice for 1 and 4 weeks as an acute and chronic fibrosis model, respectively. Along 1st to 4th weeks; vitamin D were i.p injected/2x week. Liver were assessed histologically and for proteins quantification for VDR and fSMA expressions. In vitro, potential killing of NK cells were evaluated following coculture with primary-hepatic-stellate-cells (pHSCs) obtained from BALB/c WTmice.
Results: Hepatic-fibrosis increased along 4 weeks of CCl4 as indicated by serum ALT, aSMA expressions (P‹0.02) and histological assessments. These results were associated with increased NK1.1 activations and hypercalcemia. While vitamin D administrations delayed fibrosis of early stages, vitamin D worsen hepatic-fibrosis of late stages of CCl4. In week 4, no further activations of NK cells were seen following vitamin D injections and were associated with down-expressions of VDR (1.7 Fold, P‹0.004) indicating the inability of vitamin D to ameliorate hepatic fibrosis. In vitro, NK cells from the chronic model of CCl4 did not affect pHSCs killing and fail to reduce fibrosis.
Conclusion: Vitamin D alleviate liver NK cytotoxicity in acute but not in chronic fibrosis model due to modulations in vitamin D receptor and calcium. Hypercalcemia associated with late fibrosis may inhibited VDR levels, however, may not explain the profibrogenic effects of vitamin D.
Keywords: Hepatic fibrosis; NK cells; Vitamin D receptor; Hypercalcemia
Abbreviations
Vitamin D receptor- VDR, Hepatic stellate cells-HSCs, Carbon tetrachloride-CCl4, 25-(OH) D3 -Vitamin D.
Introduction
Vitamin D is an important prohormone with known effect on calcium homeostasis [1], but recently there is increasing recognition that vitamin D also is involved in cell proliferation and differentiation, it has immunomodulatory and anti-inflammatory properties [2]. The effects of vitamin D are mediated through the vitamin D receptor (VDR) [3]. VDR is a member of the nuclear receptor super-family of ligand-inducible transcription factors, which are involved in many physiological processes, including cell growth and differentiation, embryonic development and metabolic homeostasis [4]. The transcriptional activity of this receptor is modulated by several ligands, such as steroids, retinoids and other lipidsoluble compounds, and by nuclear proteins acting as co-activators and co-repressors [5]. The liganded VDR heterodimerizes with the retinoid X receptor and binds to vitamin D response elements in the promoter of target genes, thereby affecting their transcription. The genomic organization of the VDR at locus 12q13.1 shows that the VDR gene itself is quite large (over 100kb) and has an extensive promoter region capable of generating multiple tissue-specific transcripts [6].
Clinical observations have recently demonstrated that 25-OH D serum levels were significantly lower in patients with chronic hepatitis C than in controls and that low 25-OH D serum levels were associated with more severe fibrosis and lower responsiveness to interferonbased therapy in those patients [7,8]. Other studies have shown that low 25-OH D serum levels are associated with poor liver function and more advanced stages of liver fibrosis in hepatitis C virus patients [9]. Moreover, a possible role for vitamin D in liver fibrosis has gained further support from the finding that VDR is expressed in human as well as in rat liver nonparenchymal cells, such as Hepatic stellate cells (HSCs) [10]. It has recently been suggested that VDR polymorphism is associated with primary biliary cirrhosis [11]. Vitamin D deficiency is a common phenomenon in chronic liver disease, particularly in advanced fibrosis and cirrhosis [12]. Whether this association reflects the cause of accelerated fibrosis progression or the consequence of impaired liver function in advanced disease is still unclear.
Materials and Methods
Animals
Male mice on the BALB/c background, 12 weeks of age, weighing 22 U± 0.5 g, received care according to NIH guidelines. Mice were purchased commercially from Harlan Laboratories, Jerusalem-Israel. All animal protocols were approved by the institutional animal care ethical committee of the Hebrew University and housed in a barrier facility.
CCl4 Model of Liver Injury and Fibrosis
BALB/c mice were IP injected with 0.5ml/kg body weight CCl4 (1:10 v/v in corn oil from Sigma) or vehicle (corn oil) twice a week for 4 weeks. 25(OH) D3 (Biogems, Cat# 3220632-10mg) at the dose of 0.5 microgram/100g body was IP injected twice a week, commencing one day after the first dose of CCl4. The animals were terminated 72hr after the final CCl4 injection, and whole livers and serum were collected for histological, cytological and biochemical analyses.
Alanine aminotransferase (ALT)
Blood samples were collected from BALB/c male hearts at the volume of 1ml blood; the samples were centrifuged for 5min at 4000rpm. Serum samples were drawn to an Eppendorf tubes, after blood centrifuge. Serum samples of 32μl were dropped onto the strip of the ALT (Reflotron Company) and analyzed by Reflovet ® plus Roche.
Liver NK cells isolation
Under deep ether anesthesia, mice were euthanized by isoflurane, USP 100% (INH), then the liver was removed and a part of the liver was transferred to Petri dish that contains 5ml DMEM medium (Biological industries; Cat# 01-055-1A). The liver tissue was thoroughly dissected by stainless steel mesh, the cells were harvested with the medium and added to 50ml tubes that contain 10ml DMEM, and then carefully cells were transferred to new tubes that contain Ficoll (Abcam; Cat# AB18115269). Tubes were centrifuged for 20 minutes, at 1,600rpm at 20°C. The supernatant in each tube was transferred to a new tube, for another centrifuge for 10 min, at 1,600 rpm at 4°C. After the second centrifuge, the pellet in each tube was resuspended in 1ml of DMEM for NK isolation kit (Stem Cells; Cat# 19665).
Primary HSCs isolation
HSCs were isolated from 12-week-old male BALB/c mice by in situ pronase, collagenase perfusion, and single-step Histogenz gradient as previously reported (Hendriks et al., 1985; Knook et al., 1982). Isolated HSCs were cultured in Dulbecco’s modified Eagle’s medium (Mediatech) containing 20% fetal bovine serum (Hyclone) on six-well plates for 40hr prior to end-point assays.
Flow cytometry
Harvested mice liver NK cells were adjusted to 106/ml in buffer saline containing 1% bovine albumin (Biological Industries; Cat# 02-023-5A), and were stained for the following antibodies: Antimice NK1.1 (murine NK cell marker) (Biogems; Cat# 83712-70), Anti CD49a (MACS; Lot# 5150716246), and anti-mice Lysosomalassociated Lysosomalassociated membrane protein-1 (CD107a; NK1.1 cells cytotoxicity marker) (eBioscience; Cat# 48-1071). All antibodies were incubated for 45min at 4°C. The cells were washed with 0.5ml of staining buffer and fixed with 20ml of 2% paraformaldehyde. All stained cells analyzed with a flow cytometer (The BD LSRFortessa™, Becton Dickinson, Immunofluorometry systems, Mountain View, CA).
Real Time PCR
Tissue RNA isolation: Total cellular RNA was isolated from liver tissue, using 2ml TRI Reagent (Bio LAB; Cat# 90102331) for each cm3 of tissue. The samples were homogenized for 5min at room temperature of 25°C. Chloroform at volume of 0.2ml (Bio LAB; Cat# 03080521) were added to each sample, incubated for 15min at room temperature and centrifuged (1,400rpm) for 15min at 4°C. RNA precipitation: The supernatant in each sample was transferred to new Eppendorf, 0.5ml of isopropanol (Bio LAB; Cat# 16260521) left for 10min at 25°C and centrifuged (12,000rpm) for 10min at 4°C. The supernatants were removed and one ml of ethanol 75% were added to the pellet and centrifuged (7,500rpm) for 5min. The pellets were dried in air at room temperature for 15min, 50μl of DEPC were added, and the samples were heated for 10min at 55°C.
cDNA preparation: Liver RNA was extracted as described above. Preparing of c-DNA was performed using High Capacity cDNA Isolation Kit (R&D; Cat # 1406197).
Real time PCR: Real-time PCR is performed for the quantification of the expression of the gene that encoded alpha Smooth Muscle Actin (aSMA) and Vitamin D Receptot (VDR), compared to GAPDH as a housekeeping gene by using Taqman Fast Advanced Master Mix (Applied Biosystem; Cat # 4371130).
Western blot analysis
Immunoblot analysis of aSMA in liver extracts performed. Following isolation, whole protein extracts were prepared in liver homogenization RIPA buffer (Sigma; R0278-50ML) with protease phosphatase inhibitor cocktail (Roche; 1183617011). Next, proteins (30μg per lane) were resolved on a 10% (wt/vol) SDS-polyacrylamide gel (Novex, Groningen, The Netherlands) under reducing conditions. For immunoblotting, proteins transferred to a PVDF membrane. Blots incubated 1h at 4°C temperature at in a blocking buffer containing 5% skim milk and then incubated with either mouse Mouse Anti Human\Mice a-SMA (Novusbio; NB600-531), Rabbit Anti-Collagen I (ab34710), Rabbit Anti-VDR (ab109234) and Mouse Anti Human\Mice β-Actin (R&D System; 937215), diluted 1/1000, overnight in 4°C, and subsequently, with peroxidase-conjugated goat anti-mouse and Rabbit IgG (PARIS, Compiegne, France) diluted 1/5,000, for 1.5h at room temperature. Immunoreactivity revealed by enhanced chemiluminescence using an ECL Kit (Abcam; ab133406).
Histological assessments of liver injury
The posterior one third of the liver was fixed in 10% formalin for 24 hours and then paraffin-embedded in an automated tissue processor. Seven-millimeter liver sections were cut from each animal. Sections (15mm) were then stained in 0.1% sirius red F3B in saturated picric acid as well as Masson trichrome stain for connective tissue stain (both from Sigma). Sirius red staining assessed using the modified Histological Activity Index (HAI) criteria, incorporating semi quantitative assessment of periportal/periseptal interface hepatitis (0- 4), confluent necrosis (0-6), focal lytic necrosis/apoptosis and focal inflammation (0-4), portal inflammation (0-4), and architectural changes/fibrosis and cirrhosis (0-6).
Statistical analysis
The results were evaluated using the Student’s t-test, with statistical significance set at P‹0.05. Comparison between the mean values of different experiments was carried out. All data were reported as mean ± SE.
Results and Discussion
25-(OH) D3 (vitamin D) reduced liver fibrosis and inflammation in acute while worsen fibrosis in chronic model of CCl4
Hepatic fibrosis was induced in BALB/c mice by biweekly IP CCl4 injections for 1 week (acute model) and 4 weeks (chronic model) and was compared with naive vehicle-treated mice (WT). Histological staining, ALT levels and aSMA expressions were performed for fibrosis assessments. Figure 1A shows staining of Trichrome and Sirius red for the three major animal groups with and without IP administration of vitamin D as described in materials and methods. Acute and chronic model of CCl4 showed gradual extent of collagenous connective tissue fibers of red fibrosis septae with both Trichrome and Sirius red stains in consistent with CCl4 exposure time.
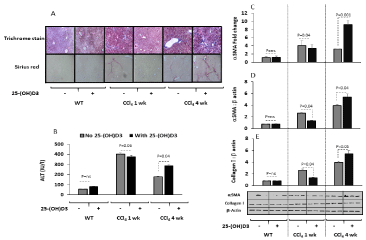
Figure 1: Vitamin D alleviates hepatic fibrosis in acute model of CCl4 while
worsen in the chronic model.
(A) Liver histology assessments showed that vitamin D IP administration (0.5
mg/100g body weight) caused a lack of hepatic fibrosis following staining with
by the Trichrome and Sirius red in the 1 week injections of CCl4 (acute model)
while aggravate collagen formations in the 4 weeks of CCl4 (chronic model).
(B) Serum ALT levels were significantly (P=0.04) increased in the acute and
chronic models of CCl4 and a further increased in ALT were seen following
vitamin D treatments only in the chronic model (p=0.04). Liver primary hepatic
stellate cells (C) RNA aSMA quantitation by RT PCR and western blot
protein quantitation of (D) aSMA and (E) Collagen showed similar patterns
to histology assessments.
Fibrotic mice of WT and acute model of CCl4 treated with vitamin D showed attenuation in their fibrosis histology that lack red fibrosis septae. On the other hand, vitamin D treatments in the chronic CCl4 model, in the contrary, had propagate liver fibrosis as seen in liver histological samples (Figure 1A).
To further evaluate effects of vitamin D on liver injury, serum ALT levels and liver samples for quantitation of fibrosis were performed. Figure 1B shows serum ALT levels were significantly (P=0.05) increased from 33.1±7.5 in naïve WT mice to 401.5±45 IU/L and 178±21 in acute and chronic model of CCl4, respectively. While vitamin D treatments had no effects on WT mice, it decreased ALT levels in acute model of CCl4 to 380±29.4 IU/L (p=0.04) and caused elevations in ALT levels in the chronic model of CCl4 up to 288.5±30.5 IU/L (P‹0.05). Gene expressions of liver aSMA fibrotic marker by RT PCR (Figure 1C), aSMA liver portion expressions by western blot analysis (Figure 1D) and liver collagen quantitation (Figure 1E) showed similar patterns to ALT. The above results showed dual antiand pro- fibrotic effects of vitamin D could be related to extent of fibrosis stage.
Liver vitamin D receptor (VDR) expressions were inhibited with fibrosis progressions and were unresponsive to vitamin D treatments
The vitamin D hormone, 1,25-dihydroxyvitamin D(3) [1,25(OH) (2)D(3)], binds with high affinity to the nuclear vitamin D receptor (VDR), and is considered as primary regulator of calcium (Ca+2). We aimed to evaluate Ca+2, vitamin D and VDR changes in our mice model with and without vitamin D treatments. Figure 2A shows similar Ca+2 serum levels in our mice model prior to vitamin D treatments. Vitamin D treatments did not change Ca+2 serum levels in WT and acute model of CCl4, however, it did significantly elevated Ca+2 in the chronic model of CCl4. In Figure 2B, vitamin D serum levels were significantly elevated in the acute model of CCl4 and were inhibited in the chronic model to levels similar to WT mice. Vitamin D treatments while did not alter vitamin D serum levels in the WT and chronic model of CCl4, it significantly dropped vitamin D serum levels 4.6 fold (P=0.004) in the acute model.
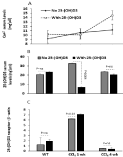
Figure 2: Chronic CCl4 mice model was associated with decreased pHSCs
VDR and hypercalcemia. (A) High Ca+2 serum levels were obtained following
vitamin D treatments only on the chronic model (p‹0.05). (B) A significant
drop in serum vitamin D levels were seen in acute mice model of CCl4
following vitamin D treatments while no changes were seen in the chronic
model. (C) Inhibition of VDR in pHSCs in the chronic model of CCl4 while upregulation
of the VDR were obtained in acute model by western blot.
Therefore, our next aim were whether changes in serum vitamin D are related to modulations in the VDR. For this purpose, primary hepatic stellate cells (pHSCs) obtained from livers of mice were quantitated for VDR by the western blot analysis. Figure 2C shows upregulation in the expressions of VDR in acute model of CCl4 as compared to the WT, while total loss of VDR expressions were seen in the chronic model of CCl4 and indicate that VDR expressions were modulated along the course of the liver injury. Vitamin D treatments significantly caused increased in VDR expressions in acute model of CCl4 (P=0.03) while had no effects in VDR expressions in the WT and the chronic model of CCl4.
The above results indicate increased Ca+2 serum levels in chronic model of CCl4 treated with vitamin D could be as a result of liver injury and in particular due to degradation of vitamin D in these mice. Consequently, these effects were associated with inhibited VDR and therefore, unlike the acute model, chronic mice model of CCl4 were unresponsive to vitamin D treatments (Figure 2B and 2C). Moreover, in the acute model of CCl4 due to the upregulation of VDR, vitamin D treatments were up taken/consumed explaining their low vitamin D levels in their serum and calcium serum levels that were unchanged.
In consistent to our data, vitamin D deficiency has been frequently reported in many causes of chronic liver disease and has been associated with the development and evolution of non-alcoholic fatty liver disease (NAFLD) [12] and chronic hepatitis C (CHC) virus infection [9].
NK cells have VDR and it is crucial for their stimulations and modulation of liver fibrosis
We next sought to determine effects of vitamin D on immune alterations in liver fibrosis. We previously showed that NK cells exert anti-fibrotic effects through killing of activated HSCs. Liver resident NK cells (NK1.1/CD49+) cellular infiltrates as well as their expressions of VDR and their activity (CD107a) were determined by the flow cytometry. Figure 3A shows an linear correlation between NK cells infiltrates with the progression of liver fibrosis. Vitamin D caused an additional increase in the NK1.1 cells infiltrates (p=0.01) only in the acute model of CCl4 that was not seen in the WT and the chronic model of CCl4 indicating a possible fibrosis inhibition in this model that were also emphasized in our precious data summarized in Figure 1 and Figure 2. VDR expressions on liver NK1.1 increased in the acute while inhibited in the chronic model of CCl4 to levels similar to WT. The VDR were upregulated following treatments with vitamin D in the WT and acute model of CCl4. No changes in the VDR were noticed in the NK1.1 cell from chronic model of CCl4 following the vitamin D treatments (Figure 3B). These data suggest inverse relation between NK1.1 cell proliferation (Figure 3A) and their VDR expressions (Figure 3B) prior and following vitamin D treatments.
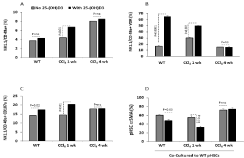
Figure 3: Increased resident liver NK 1.1/CD49a+ cells in the chronic model
of CCl4 is correlated with their dysfunction and low VDR expressions.
(A) Liver NK 1.1/CD49a+ cells showed gradual elevations in proliferations
together with the extent of hepatic fibrosis; vitamin D treatments showed
further liver NKs recruitments. (B) NK 1.1/CD49a+ were responsive to vitamin
D treatments through increase in their VDR in the WT and acute CCl4 model
however were unresponsive in the chronic model. (C) NK 1.1/CD49a+ showed
similar CD107a in the vitamin D untreated mice with further stimulation in the
WT and acute model of CCl4. (D) NK1.1 cells from the WT and acute model of
CCl4 showed to inhibit pHSCs aSMA percentages following co-culture assay
following vitamin D treatments. NK1.1 cells from the chronic model of CCl4
showed no effects on aSMA percentages of pHSCs.
To further correlate whether changes in the VDR on NK1.1 cells is associated with their phenotype alterations; CD107a (activity marker) was determined by the flow cytometry as described in materials and methods. CD107a NK1.1 percentages were high in both CCl4 models to similar levels of the WT. Vitamin D treatments further elevated NK1.1 cells activity in the WT and the acute model of CCl4 (P‹0.05) while did not alter NK1.1 activities in the chronic model most probably because the VDR is low in the chronic model of CCl4. In order to determine whether changes in CD107a and VDR on NK1.1 cells could modulate liver fibrosis; we co-cultured liver NK1.1 cells with pHSCs through an in vitro setting assay to assess cytotoxicity/ killing potential. Figure 3D shows NK1.1 cells obtained from the acute model of CCl4 reduced aSMA percentages of pHSCs as compared to their WT counterparts, a result that indicate killing of pHSCs. NK1.1 cells from the chronic model of CCl4 did not change aSMA of the pHSCs indicating inability of these cells to reduce fibrosis. Vitamin D treatments further exacerbate NK1.1 killing potentials in both the WT and the acute model and inhibited fibrosis while did not cause any changes in aSMA of the pHSCs in the chronic model of CCl4.
The above results are associated with VDR lost in NK1.1 cells from the chronic model of CCl4 that attenuated their potentials to kill. HSCs play a critical role in apoptosis and inflammation [10]. Activated HSCs secrete pro-fibrogenic cytokines, produce aSMA and pro-collagen I, and are known to be a major source of collagens and inhibitors of matrix-degrading enzymes (tissue inhibitor of matrix metalloproteinase, TIMP) that are secreted during fibrosis [10].
Studies show that hepatocytes isolated from humans, rats, and mice express little or no VDR mRNA and protein [13,14]. On the other hand, non-parenchymal cells, including HSCs, sinusoidal endothelial cells, and Kupffer cells (KCs), strongly express VDR [15], and these probably mediate vitamin D effects in the nondiseased state. However, levels of VDR in hepatocytes increase with inflammation, giving a broader site for potential targets. Levels also increase in HSCs and KCs with inflammation, and several immune functions of VDR described in other tissues may be involved in liver disease.
References
- Norman AW, Roth J, Orci L. The vitamin D endocrine system: steroid metabolism, hormone receptors, and biological response (calcium binding proteins). Endocr Rev. 1982; 3: 331-366.
- Deluca HF. Historical overview of Vitamin D. In: Feldman D, Pike JW, Adams JS, editors. , eds. Vitamin D. Vol 1 3rd ed Boston, MA: Elsevier. 2011: 3-12.
- Mizwicki MT, Norman AW. The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling. Sci Signal. 2009; 2: 4.
- DeLuca HF. Evolution of our understanding of vitamin D. Nutr Rev. 2008; 66: 73-87.
- Haussler MR, Whitfield GK, Kaneko I, et al. Molecular mechanisms of vitamin D action. Calcif Tissue Int. 2013; 92: 77-98.
- Ingraham BA, Bragdon B, Nohe A. Molecular basis of the potential of vitamin D to prevent cancer. Curr Med Res Opin. 2008 24: 139-149.
- Palazzo D, Biliotti E, Esvan R, Volpicelli L, Franchi C, Fontanelli Sulekova L, et al. Vitamin D deficiency and health-related quality of life in chronic hepatitis C. J Viral Hepat. 2019; 26: 774-777.
- Gayam V, Mandal AK, Khalid M, Mukhtar O, Gill A, Garlapati P, et al. Association Between Vitamin D Levels and Treatment Response to Direct- Acting Antivirals in Chronic Hepatitis C: A Real-World Study. Gastroenterology Res. 2018; 11: 309-316.
- Jin CN, Chen JD, Sheng JF. Vitamin D deficiency in hepatitis C virus infection: what is old? what is new? Eur J Gastroenterol Hepatol. 2018; 30: 741-746.
- Udomsinprasert W, Jittikoon J. Vitamin D and liver fibrosis: Molecular mechanisms and clinical studies. Biomed Pharmacother. 2019; 109: 1351- 1360.
- Fang F, Wang J, Pan J, Su GH, Xu LX, Li G. Relationship between vitamin D (1, 25-dihydroxyvitamin D3) receptor gene polymorphisms and primary biliary cirrhosis risk: a meta-analysis. Genet Mol Res. 2015; 14: 981-988.
- Arai T, Atsukawa M, Tsubota A, Koeda M, Yoshida Y, Okubo T, et al. Association of vitamin D levels and vitamin D-related gene polymorphisms with liverfibrosis in patients with biopsy-proven nonalcoholic fatty liver disease. Dig Liver Dis. 2019; 51: 1036-1042.
- Bookout A.L., Jeong Y., Downes M., Yu R.T., Evans R.M., Mangelsdorf D.J. Anatomical Profiling of Nuclear Receptor Expression Reveals a Hierarchical Transcriptional Network. Cell. 2006; 126: 789-799.
- Han S., Li T., Ellis E., Strom S., Chiang J.Y.L. A Novel Bile Acid-Activated Vitamin D Receptor Signaling in Human Hepatocytes. Mol. Endocrinol. 2010; 24: 1151-1164.
- Gascon-Barré M., Demers C., Mirshahi A., Néron S., Zalzal S., Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology. 2003; 37: 1034-1042.