
Review Article
Austin J Genet Genomic Res. 2014;1(1): 5.
Age Related Macular Degeneration: a Complex Pathology
Sandeep Kumar1* and Yingbin Fu1,2
1Department of Ophthalmology & Visual Sciences, University of Utah School of Medicine, USA
2Department of Neurobiology & Anatomy, University of Utah Health Sciences Center, USA
*Corresponding author: Sandeep Kumar, Department of Ophthalmology and Visual Sciences, University of Utah School of Medicine, John A. Moran Eye Center, 65 Mario Capecchi DriveSalt Lake City, Utah 84132, USA
Received: June 20, 2014; Accepted: July 22, 2014; Published: July 23, 2014
Abstract
Age-Related Macular Degeneration (AMD) is the leading cause of severe irreversible central vision loss and blindness in individuals of over 65 years of age in the developed countries. There are two types of AMD, the ''dry'' and ''wet'' forms. Both genetic and non-genetic (environmental) factors are considered for the onset of AMD. The etiology and pathogenesis of AMD are not well understood and remain a major challenge to understand. This review discusses recent advancement in genetics and genomics, and the molecular pathways involved in AMD pathogenesis.
Keywords: Age-Related Macular Degeneration; Choroidal Neovascularization; Polypoidal Choroidal Vasculopathy; HTRA1; Complement Factor H; Inflammation; Autophagy
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness worldwide. At present, 8•7% of the worldwide population has AMD and by 2010 around 196 million expected to have AMD which is further increasing to 288 million in 2040 [1]. There are two types of AMD, the ''dry'' and ''wet'' forms. Wet AMD includes classic and occult choroidal neovascularization (CNV) and polypoidal choroidal vasculopathy (PCV). The chronic form "Dry AMD" typically develops first and is characterized by the deposition of acellular, polymorphous debris between the retinal pigment epithelium (RPE) and Bruch's membrane (BM) called "drusen". The excessive "drusen" deposition may lead to damage of the RPE, degeneration of collagen or elastin in BM, the outer retina and the choroid vasculature, which may lead to wet form of AMD where abnormal vessels grow within the sub-RPE space or grow out in to the retina by rupturing the RPE, this form occurs at the late stage. Therefore, dry AMD is considered a precursor for the wet AMD. Caucasian AMD patients predominantly exhibit late stage geographic atrophy of dry "AMD" while Asian AMD patients frequently have CNV or PCV forms of "wet AMD" with few or no drusen. Wet AMD represents only 10 to 15% of the overall prevalence of AMD but is responsible for more than 80% of cases of legal blindness [2]. Overall, AMD is a progressive, polygenic and multi factorial disease with a poorly understood etiology. Numerous studies have suggested the involvement of advanced age, race, heredity, and a history of smoking and alcohol drinking, oxidative stress, inflammation and immune response [3,4], which makes AMD pathology extremely complex.
AMD Genetics
Over the years, the involvement of genetics in the development of AMD has been very well studied and established. Genome-Wide Association Studies (GWAS) have revealed more than 30 risk loci ( e.g. 1q25-31, 9p13, 9p24, 10q26, 15q21, and 17q25) and have implicated several candidate genes-CFH, C3, C2-CFB, CFI, HTRA1/ ARMS2, CETP, TIMP3, LIPC, VEGFA, COL10A1, TNFRSF10A, and APOE with AMD [5-7]. Among them, chromosome loci 1q32 and 10q26 are major candidate regions associated with the susceptibility of AMD [5,8-17] including PCV [18-20]. The genetic variants in complement factor H at chromosome loci 1q32 and additional complement-related genes firmly established a link between the complement cascade and AMD biology, which have been implicated in mediating drusen formation [21]. CFH Y402H is a major AMD susceptibility variant in Caucasians and has been shown that heterozygote alleles conferred a 4.6-fold where-as homozygote alleles have a 7.4-fold increased risk, as compared with the homozygous non-risk genotype [14]. On the other hand, chromosome loci 10q26 is more complex due to the strong linkage disequilibrium (LD) across this region comprising of three genes: pleckstrin homology domain containing family A member 1 (PLEKHA1), age-related maculopathy susceptibility 2 (ARMS2) and high-temperature requirement A serine peptidase 1 (HTRA1) [8,14-17]. Because of strong LD, statistical genetic analysis alone is incapable of distinguishing the effect of an individual gene in this locus and has yielded widely conflicting results [15,17,22- 28]. As a result, the functional involvement of HTRA1, ARMS2 or PLEKHA1 in AMD remains uncertain, despite strong genetic evidence. So far, rs10490924, indel polymorphisms of ARMS2, and rs11200638 of HTRA1 promoter region are most significantly AMD associated haplotypes at this locus [29]. The HTRA1 gene encodes an evolutionarily conserved multifunctional serine protease that is ubiquitously expressed in mammalian tissues but ARMS2 is only expressed in certain primates with unknown function. The sub cellular localization of ARMS2 is controversial and studies suggesting that it present in mitochondria, extracellular matrix, or as a non coding RNA [16,17,27]. An increased level of HTRA1 is suggested to play a potential role in the pathogenesis of AMD [15,23-25]. Therefore, we studied the functional involvement of HTRA1 by transgenically expressing human HTRA1 in mouse RPE and showed that increased HTRA1 induced characteristic features of PCV, including branching networks of choroidal vessels (BVN) and polypoidal lesions (polyps). Ultra structural study revealed degeneration of both the elastic lamina and tunica media of choroidal vessels, as well as the degradation of the elastic lamina of Bruch's membrane in hHTRA1+ mice. Another group also reported the degradation of EL in BM when over expressing mouse HTRA1 in RPE [30]. The phenotypes of hHTRA1+ mouse we generated share remarkable similarities to the well established clinical features of human PCV (e.g. BVN, polyps, late geographic hyper fluorescence, pigment epithelium detachment, and hyper fluorescent plaque) [31-33]. The hHTRA1+ mouse is the first PCV model and no other animal models exist with these features. The strengths and limitations of available AMD animal models are comprehensively reviewed by Pennesi ME [34]. HTRA1 is clearly important in maintaining the vasculature by inhibiting the signaling of TGFb family members [35,36]. Loss-of-function mutations in HTRA1 were linked to familial ischemic cerebral small-vessel disease [37,38]. In the eye, knockout of HTRA1 leads to reduced blood vessels in mouse retina [39]. However, several studies demonstrated that AMD associated variants at 10q26 locus are not correlated with the expression level of HTRA1 in AMD-affected eyes [26, 27,40- 42]. Recently, it is shown that AMD linked synonymous SNPs within exon 1 of HTRA1 makes it conformationally defective. This conformationally defective HTRA1 is more susceptible to proteolysis and has a reduced binding capacity to IGF-1, which supports cellular division and growth therefore may compromise photoreceptors and choriocapillaris survival [43]. Currently, all three possibilities (up-regulation, down-regulation or no change) in HTRA1 levels with AMD-associated variants are being investigated. HTRA1 is the leading candidate for the 10q26 genetic risk. However, more studies are necessary to establish a firm link.
Inflammation and AMD
In recent years, numerous clinical-genetic studies documented the crucial role of inflammation and immune-mediated processes (e.g. complement activation) in the pathogenesis of AMD. The ectopic levels of complement components C3a and C5a, C5 and C5b-9 terminal complement complex [44-46], complement factor H (CFH) [13, 47], membrane cofactor protein (MCP) [48], and C-reactive protein (CRP) [49] are observed in AMD patients and clearly indicating that complement activation is crucial in AMD pathogenesis. In fact, the hallmark of AMD, "drusen", contains large amount of components involved in the complement pathway [44,50- 57]. In addition, it's been shown that Membrane Attacking Complex (MAC) formation is increased in the photoreceptors that may trigger the apoptotic processes inducing retinal degeneration [50-53,57- 58]. The deposition of esterified/unesterified cholesterol (7kCh) and glycation/lipoxidation end products (AGEs/ALEs) has been identified in the retina, BM and in RPE/choroid of human AMD donor eyes [59-61], suggesting that lipid metabolism pathways also have a crucial role in AMD pathogenesis via inflammation. The accumulation of macrophages in the AMD tissues suggest an important role for macrophages in AMD pathogenesis [62-65], which is well supported in AMD animal model studies [66-70]. However, macrophage populations are heterogeneous and can be both protective and destructive to local tissues. Based on macrophage functions, surface markers, and cytokine/chemokine profiles they are characterized as classically activated macrophages (M1), which are generally pro-inflammatory. On the other hand, alternatively, activated macrophages (M2), facilitate tissue repair and neovascularization. Both types of macrophages have been characterized in only a limited number of AMD tissues samples [62,63]. The precise roles and impacts of macrophages in AMD are unclear and debated in the AMD field. It is important that more histo chemical studies shall be performed to elucidate those factors that alter macrophage polarity and mediate angiogenesis. These factors may have the potential of aiding in new anti-inflammatory therapies for AMD. Recent studies suggest NLRP3 inflammasome may play a critical role in AMD [71]. NLRP3 is an intracellular pattern-recognition receptor, which responds to a wide variety of danger signals. The exact mechanism by which NLRP3 inflammasomes become activated has remained unclear. During the past decade, the major breakthrough is the development of anti-VEGF therapy for wet AMD. VEGF-A induces proliferation, sprouting and tube formation of endothelial cells and plays a major role in CNV. In addition to VEGF, aberrant levels of interleukins IL-6, IL-8 and IL-10 are also found in - CNV patients.
Autophagy and AMD
Recently, autophagy has caught the attention of AMD researchers. Autophagy plays a critical role in removing misfolded or aggregated proteins, clearing damaged organelles, such as mitochondria, endoplasmic reticulum and peroxisomes [72]. It also eliminates intracellular pathogens to keep post-mitotic cells healthy and functional [73]. The autophagy processes are highly active in the RPE layer because RPE cells are subject to oxidative stress, high oxygen tension, lifelong light illumination, and are involve in daily phagocytosis of photoreceptor outer segments. As we read in the previous section, the physiological balance between various interlinked pathways (eg vascular growth factor pathways, lipid pathways and oxidative stress pathways) has been perturbed in AMD which may impair the autophagy process. Some exosome and autophagy markers have been detected in drusen [74]. Inflammation and local hypoxia are the hallmarks of autophagy and are present in the aging choriocapillaris, RPE cells, and neural retina [75]. It is well known that oxidative stress leads to mitochondrial DNA damage, increases ROS generation and reduces the metabolic capacity. The increased mitochondrial stress and dysfunctional autophagy in the RPE cells of AMD patients also support the involvement of autophagy in the pathology of AMD [76,77]. The association between the variant of CST3, (encoding cystatin C), an inhibitor of lysosomal cysteine proteases, and AMD has been established. Also, increased serum levels of cystatin C found in AMD patients are correlated with the risk of development of advanced AMD [78,79]. In addition, in-vitro studies on lysosome function on RPE cells also provided insights on the disruption of lysosomal functions and possible role of lysosomes in the development of AMD [79-82]. Vascular dysfunctions also result in oxidative stress, that is, overproduction of ROS, which induces further changes in the retinal and choroidal vasculature. Such changes can also be evoked by hypoxia, since it stimulates synthesis and release of hypoxia-inducible factor-1 (HIF- 1) and vascular endothelial growth factor (VEGF) that contribute to neovascularization (NV). Recent reports suggest that dysfunctional autophagy activates inflammasomes probably through the dysregulation of mitochondrial homeostasis [83,84]. To date, there is no consensus as to whether autophagy inhibitors or activators would be beneficial in AMD therapy.
Treatment
There is no cure for AMD. Nevertheless, AMD treatment may prevent severe vision loss or slow the progression of the disease considerably, for example, anti-angiogenic drugs (anti-VEGF) and photodynamic therapy with verteporfin (PDT-V) are very effective for wet AMD. However, the anti-VEGF therapy is not very effective in treating PCV compared with classic CNV (or type 2 neovascularization). Monoclonal antibodies Ranibizumab (Lucentis) and Bevacizumab (Avastin) are used to treat "wet form" of AMD by targeting all isoforms of VEGF-A. Currently, bevacizumab is the most widely used anti-VEGF agent throughout the world due to its significantly lower cost and similar efficacy compare to Lucentis. Another new promising drug is Aflibercept (known as VEGF-Trap) is a human recombinant fusion protein which consists of extracellular domains of VEGF receptor 1 and 2 (VEGFR-1 and -2) fused with the Fc portion of IgG1. It binds to VEGF-A, VEGF-B, and placental growth factor (PlGF). It has a higher affinity for VEGF compared to other anti-VEGFs, including bevacizumab and ranibizumab. For more detail we recommend reading the recent review from Hanout et al. 2013 [85]. Indeed, currently very little is available to prevent the progression to more serious stages for "dry" AMD's patients. Colloquially, quitting smoking and a healthy diet of dark green leafy vegetables and fruits supplemented by zinc and anti-oxidant vitamins (Vitamins E, C, and beta carotene) are recommended.
Conclusion
AMD is a genetically well-characterized disease with a high complexity. Despite several important findings in the last decade, we still do not have a clear picture of biological pathways that are actual culprits for AMD. Based on recent findings, the dysfunction and/or degenerative damages photoreceptors, RPE and BM of the macula, are initiated by "attacks" from drusen, aging, genetic and environmental risk factors. These primary factors create a para-inflammatory environment which may provoke the infiltration of macrophages, lymphocytes, neutrophils and various cytokines to the degenerated tissue sites in AMD patients and cause further damage and lead to "wet AMD" (Figure1). We have witnessed remarkable progress in identifying genetic risk factors for AMD. However, investigations of the underlying disease mechanisms by causal alleles are needed. It is also important to elucidate factors and/or signaling pathways that regulate inflammation, oxidative stress, and autophagy of this disease in order to develop effective preventive and treatment therapies.
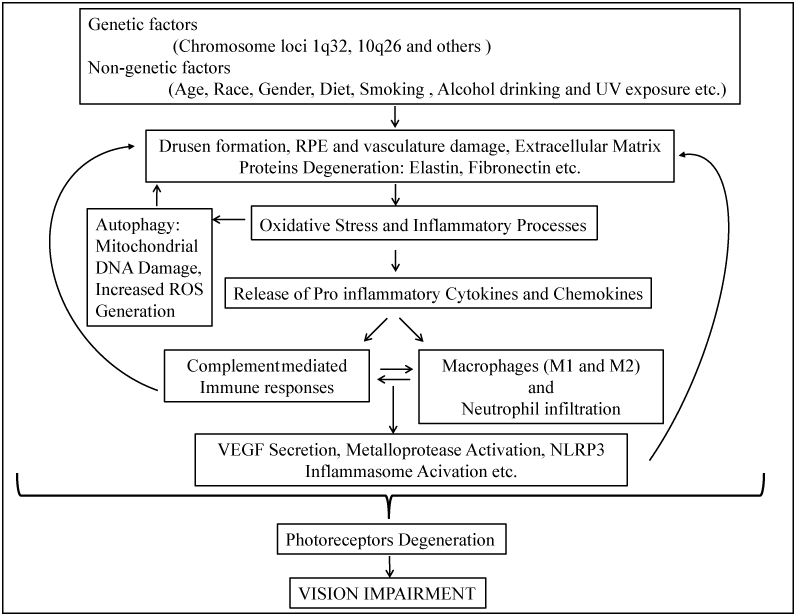
Figure 1 : A schematic representation on the risk factors and possible pathological mechanisms of AMD.
Acknowledgement
This work was supported by NIH grant 1R01EY022901, P30 EY014800, the Career Development Award from Research to Prevent Blindness (RPB), C. M. Reeves & M. A. Reeves Foundation, E. Matilda Ziegler Foundation for the Blind, Knights Templar Eye Foundation, and an unrestricted grant to the Department of Ophthalmology at the University of Utah from RPB.
References
- Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng Y, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. The Lancet Global Health. 2014; 2: 106-116.
- Wong TY, Chakravarthy U, Klein R, Mitchell P, Zlateva G, Buggage R, Fahrbach K. The natural history and prognosis of neovascular age-related macular degeneration: a systematic review of the literature and meta-analysis. Ophthalmology. 2008; 115: 116-126.
- Klein R, Peto T, Bird A, Vannewkirk MR. The epidemiology of age-related macular degeneration. Am J Ophthalmol. 2004; 137: 486-495.
- Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421-424.
- Majewski J, Schultz DW, Weleber RG, Schain MB, Edwards AO, Matise TC, et al. Age-related macular degeneration--a genome scan in extended families. Am J Hum Genet. 2003; 73: 540-550.
- Fisher SA, Abecasis GR, Yashar BM, Zareparsi S, Swaroop A, Iyengar SK, et al. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet. 2005; 14: 2257-2264.
- Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013; 45: 433-439, 439e1-2.
- Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005; 14: 3227-3236.
- Hageman GS, Hancox LS, Taiber AJ, Gehrs KM, Anderson DH, Johnson LV, et al. Extended haplotypes in the complement factor H (CFH) and CFH related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med. 2006; 38: 592-604.
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308: 419-421.
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385-389.
- Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006; 38: 1173-1177.
- Magnusson KP, Duan S, Sigurdsson H, Petursson H, Yang Z, Zhao Y, et al. CFH Y402H confers similar risk of soft drusen and both forms of advanced AMD. PLoS Med. 2006; 3: e5.
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006; 314: 989-992.
- Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006; 314: 992-993.
- Kanda A, Chen W, Othman M, Branham KE, Brooks M, Khanna R, et al. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA, is strongly associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2007; 104: 16227-16232.
- Fritsche LG, Loenhardt T, Janssen A, Fisher SA, Rivera A, Keilhauer CN, et al. Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNA. Nat Genet. 2008; 40: 892-896.
- Kondo N, Honda S, Ishibashi K, Tsukahara Y, Negi A. LOC387715/HTRA1 variants in polypoidal choroidal vasculopathy and age-related macular degeneration in a Japanese population. Am J Ophthalmol. 2007; 144: 608-612.
- Lee KY, Vithana EN, Mathur R, Yong VH, Yeo IY, Thalamuthu A, et al. Association analysis of CFH, C2, BF, and HTRA1 gene polymorphisms in Chinese patients with polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci. 2008; 49: 2613-2619.
- Lima LH, Schubert C, Ferrara DC, Merriam JE, Imamura Y, Freund KB, et al. Three major loci involved in age-related macular degeneration are also associated with polypoidal choroidal vasculopathy. Ophthalmology. 2010; 117: 1567-1570.
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227-7232.
- Friedrich U, Myers CA, Fritsche LG, Milenkovich A, Wolf A, Corbo JC, et al. Risk- and non-risk-associated variants at the 10q26 AMD locus influence ARMS2 mRNA expression but exclude pathogenic effects due to protein deficiency. Hum Mol Genet. 2011; 20: 1387-1399.
- Yang Z, Tong Z, Chen Y, Zeng J, Lu F, Sun X, et al. Genetic and functional dissection of HTRA1 and LOC387715 in age-related macular degeneration. PLoS Genet. 2010; 6: e1000836.
- Chan CC, Shen D, Zhou M, Ross RJ, Ding X, Zhang K, et al. Human HtrA1 in the archived eyes with age-related macular degeneration. Trans Am Ophthalmol Soc. 2007; 105: 92-97.
- Tuo J, Ross RJ, Reed GF, Yan Q, Wang JJ, Bojanowski CM, et al. The HtrA1 promoter polymorphism, smoking, and age-related macular degeneration in multiple case-control samples. Ophthalmology. 2008; 115: 1891-1898.
- Kanda A, Stambolian D, Chen W, Curcio CA, Abecasis GR, Swaroop A. Age-related macular degeneration-associated variants at chromosome 10q26 do not significantly alter ARMS2 and HTRA1 transcript levels in the human retina. Mol Vis. 2010; 16: 1317-1323.
- v
- An E, Sen S, Park SK, Gordish-Dressman H, Hathout Y. Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis Sci. 2010; 51: 3379-3386.
- Wang G. Chromosome 10q26 locus and age-related macular degeneration: a progress update. Exp Eye Res. 2014; 119: 1-7.
- Vierkotten S, Muether PS, Fauser S. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch's membrane via cleavage of extracellular matrix components. PLoS One. 2011; 6: e22959.
- Jones A, Kumar S, Zhang N, Tong Z, Yang JH, Watt C, et al. Increased expression of multifunctional serine protease, HTRA, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci U S A. 2011; 108:14578-14583.
- Kumar S, Berriochoa Z, Jones AD, Fu Y. Detecting abnormalities in choroidal vasculature in a mouse model of age-related macular degeneration by time-course indocyanine green angiography. J Vis Exp. 2014; 84:e51061. doi: 10.3791/51061.
- Kumar S, Berriochoa Z, Ambati BK, Fu Y. Angiographic Features of Transgenic Mice With Increased Expression of Human Serine Protease HTRA1 in Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci. 2014; 55: 3842-3850.
- Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med. 2012; 33: 487-509.
- Oka C, Tsujimoto R, Kajikawa M, Koshiba-Takeuchi K, Ina J, Yano M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development. 2004; 131: 1041-1053.
- Launay S, Maubert E, Lebeurrier N, Tennstaedt A, Campioni M, Docagne F, et al. HtrA1-dependent proteolysis of TGF-beta controls both neuronal maturation and developmental survival. Cell Death Differ. 2008; 15: 1408-1416.
- Hara K, Shiga A, Fukutake T, Nozaki H, Miyashita A, Yokoseki A, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009; 360: 1729-1739.
- Shiga A, Nozaki H, Yokoseki A, Nihonmatsu M, Kawata H, Kato T, et al. Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-Î21 via cleavage of proTGF-Î21. Hum Mol Genet. 2011; 20: 1800-1810.
- Zhang L, Lim SL, Du H, Zhang M, Kozak I, Hannum G, et al. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-β family member growth differentiation factor 6. J Biol Chem. 2012; 287: 1520-1526.
- Chowers I, Meir T, Lederman M, Goldenberg-Cohen N, Cohen Y, Banin E, et al. Sequence variants in HTRA1 and LOC387715/ARMS2 and phenotype and response to photodynamic therapy in neovascular age-related macular degeneration in populations from Israel. Mol Vis. 2008; 14: 2263-2271.
- Newman AM, Gallo NB, Hancox LS, Miller NJ, Radeke CM, Maloney MA, et al. Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med. 2012; 4: 16.
- Wang G, Dubovy SR, Kovach JL, Schwartz SG, Agarwal A, Scott WK, et al. Variants at chromosome 10q26 locus and the expression of HTRA1 in the retina. Exp Eye Res. 2013; 112: 102-105.
- Jacobo SM, Deangelis MM, Kim IK, Kazlauskas A. Age-related macular degeneration-associated silent polymorphisms in HtrA1 impair its ability to antagonize insulin-like growth factor 1. Mol Cell Biol. 2013; 33: 1976-1990.
- Nozaki M, Raisler BJ, Sakurai E, Sarma JV, Barnum SR, Lambris JD, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006; 103: 2328-2333.
- Sivaprasad S, Adewoyin T, Bailey TA, Dandekar SS, Jenkins S, Webster AR, et al. Estimation of systemic complement C3 activity in age-related macular degeneration. Arch Ophthalmol. 2007; 125: 515-519.
- Machalinska A, Dziedziejko V, Mozolewska-Piotrowska K, Karczewicz D, Wiszniewska B, Machalinski B. Elevated plasma levels of C3a complement compound in the exudative form of age-related macular degeneration. Ophthalmic Res. 2009; 42: 54-59.
- Lommatzsch A, Hermans P, Weber B, Pauleikhoff D. Complement factor H variant Y402H and basal laminar deposits in exudative age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2007; 245: 1713-1716.
- Fett AL, Hermann MM, Muether PS, Kirchhof B, Fauser S. Immunohistochemical localization of complement regulatory proteins in the human retina. Histol Histopathol. 2012; 27: 357-364.
- Bhutto IA, Baba T, Merges C, Juriasinghani V, McLeod DS, Lutty GA. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol. 2011; 95: 1323-1330.
- Mullins RF, Johnson LV, Anderson DH, Hageman GS. Characterization of drusen-associated glycoconjugates. Ophthalmology. 1997; 104: 288-294.
- Russell SR, Mullins RF, Schneider BL, Hageman GS. Location, substructure, and composition of basal laminar drusen compared with drusen associated with aging and age-related macular degeneration. Am J Ophthalmol. 2000; 129: 205-214.
- Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000; 70: 441-449.
- Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000; 14: 835-846.
- Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer's A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 11830-11835.
- Radu RA, Hu J, Jiang Z, Bok D. Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J Biol Chem. 2014; 289: 9113-9120.
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 14682-14687.
- Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006; 103: 16182-16187.
- Rudolf M, Malek G, Messinger JD, Clark ME, Wang L, Curcio CA. Sub-retinal drusenoid deposits in human retina: organization and composition. Exp Eye Res. 2008; 87: 402-408.
- Elner VM. Retinal pigment epithelial acid lipase activity and lipoprotein receptors: effects of dietary omega-3 fatty acids. Transactions of the American Ophthalmological Society. 2002; 100: 301-338.
- Rodríguez IR, Larrayoz IM. Cholesterol oxidation in the retina: implications of 7KCh formation in chronic inflammation and age-related macular degeneration. J Lipid Res. 2010; 51: 2847-2862.
- Beattie JR, Pawlak AM, Boulton ME, Zhang J, Monnier VM, McGarvey JJ, et al. Multiplex analysis of age-related protein and lipid modifications in human Bruch's membrane. FASEB J. 2010; 24: 4816-4824.
- Nakashizuka H, Mitsumata M, Okisaka S, Shimada H, Kawamura A, Mori R, et al. Clinicopathologic findings in polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci. 2008; 49: 4729-4737.
- Cherepanoff S, McMenamin P, Gillies MC, Kettle E, Sarks SH. Bruch's membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br J Ophthalmol. 2010; 94: 918-925.
- Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, et al. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. 2011; 61: 528-535.
- Espinosa-Heidmann DG, Suner IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW. Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003; 44: 3586-3592.
- Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J. Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2003; 44: 3578-3585.
- Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest. 2007; 117: 3421-3426.
- Cruz-Guilloty F, Saeed AM, Echegaray JJ, Duffort S, Ballmick A, Tan Y, et al. Infiltration of proinflammatory m1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. Int J Inflam. 2013; 2013: 503725.
- Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013; 17: 549-561.
- Ding JD, Kelly U, Groelle M, Christenbury JG, Zhang W, Bowes Rickman C. The role of complement dysregulation in AMD mouse models. Adv Exp Med Biol. 2014; 801: 213-219.
- Campbell M, Doyle SL. An eye on the future of inflammasomes and drug development in AMD. J Mol Med (Berl). 2013; 91: 1059-1070.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010; 221: 3-12.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132: 27-42.
- Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy. 2009; 5: 563-564.
- Arjamaa O, Nikinmaa M, Salminen A, Kaarniranta K. Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD). Ageing Res Rev. 2009; 8: 349-358.
- Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008; 49: 2848-2855.
- Lin H, Xu H, Liang FQ, Liang H, Gupta P, Havey AN, et al. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011; 52: 3521-3529.
- Zurdel J, Finckh U, Menzer G, Nitsch RM, Richard G. CST3 genotype associated with exudative age related macular degeneration. Br J Ophthalmol. 2002; 86: 214-219.
- Klein R, Knudtson MD, Lee KE, Klein BE. Serum cystatin C level, kidney disease markers, and incidence of age-related macular degeneration: the Beaver Dam Eye Study. Arch Ophthalmol. 2009; 127: 193-199.
- Chen PM, Gombart ZJ, Chen JW. Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci. 2011; 1: 10.
- Chen PM, Gombart ZJ, Chen JW. Chloroquine treatment of ARPE-19 cells leads to lysosome dilation and intracellular lipid accumulation: possible implications of lysosomal dysfunction in macular degeneration. Cell Biosci. 2011; 1: 10.
- Kaarniranta K, Kauppinen A, Blasiak J, Salminen A. Autophagy regulating kinases as potential therapeutic targets for age-related macular degeneration. Future Med Chem. 2012; 4: 2153-2161.
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469: 221-225.
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011; 12: 222-230.
- Hanout M, Ferraz D, Ansari M, Maqsood N, Kherani S, Sepah YJ, et al. Therapies for neovascular age-related macular degeneration: current approaches and pharmacologic agents in development. Biomed Res Int. 2013; 2013: 830837.