
Special Article - Genetics of Huntington’s Disease
Austin J Genet Genomic Res. 2016; 3(1): 1022.
Huntington’s Disease Associated Genes: Molecular Basis of Disease and Possible New Targets for Treatment
Bhattacharyya NP*
BioMedical Genomics Centre, India
*Corresponding author: Nitai P Bhattacharyya, BioMedical Genomics Centre, PG Polyclinics (3rd Floor), 5 Suburban Hospital Road, Kolkata 700 20, India
Received: June 22, 2016; Accepted: November 11, 2016; Published: November 18, 2016
Abstract
Diverse molecular defects in Huntington’s Disease (HD) have been identified since the discovery of Huntingtin (HTT) gene mutated in the diseasein1993. Presently, there is no cure for the disease. To identify genes involved in HD pathogenesis, we collate data from diverse sources and using stringent criteria collected 523 HD associated genes. Enrichment analysis with these proteins revealed that diverse biological processes and pathways like gene expression, apoptosis, proteasomal degradation, glucose/carbohydrate metabolism, known to involve in HD pathogenesis, were significantly enriched. HD associated genes were significantly over represented in biological processes like cell cycle and differentiation indicating their possible role in HD pathogenesis. Comparisons of HD associated genes with targets of FDA approved drugs and probable protein targets having similar properties as that of FDA approved protein targets of drugs, we identified 49 HD associated genes that could be new probable drug targets. HD associated genes collected here will be useful to decipher molecular basis of HD including regulation of these genes and new targets for the treatment of presently incurable devastating HD.
Keywords: Huntington’s disease; Huntingtin interacting proteins; Coexpressed genes; Drug targets
Abbreviations
HD: Huntington’s Disease; FDA: Food and Drug Administration; AAO: Age at Onset; TVS: Target validation score
Introduction
Huntington’s Disease (HD, OMIM ID 143100), also well known as Huntington’s chorea, is an autosomal dominant progressive degenerative neurological rare disease (Orphanet ID ORPHA399), named after George Huntington, who first provided vivid systematic descriptions of the disease [1]. Typical characteristic of HD is abrupt involuntary movement (chorea) due to random muscle contractions. Other features of HD in early stage of the disease include behavioral changes like agitation, irritability, apathy, anxiety, dis-inhibition, euphoria, delusions, hallucinations, depression, dementia, cognitive decline and motor function impairment like eye movement abnormality, parkinsonian feature, dystonia, myoclonus, ataxia, dysarthria, dysphagia and spasticity with hyperreflexia. Dystonia or akinetorigid parkinsonian features supersede chorea with progression of disease [2,3]. HD is caused by expansion of normally polymorphic CAG repeats beyond 36 at the exon1 of the gene Huntingtin (HTT), also known as IT15 [4]. Manifestation of first symptoms, defined as the Age at Onset (AAO), is variable and normally ranges between 30 and 40 years. Early onset (<20 years, Juvenile HD) and higher age at onset (~70 years) have also been reported. The disease progresses slowly and within 15-20 years leads complete dependency in regular daily activities requiring full-time care and finally death. Common cause of death among HD patients is pneumonia and suicide. Other than classical symptoms, several features, which are not associated with the neuronal loss, complicate HD. These include weight loss and skeletal muscle loss. It is unclear whether such losses are secondary to neurological dysfunctions. However, such effects may contribute to the mortality or morbidity [5]. In spite of a large number of studies, since discovery of the gene that is mutated in HD since 1993, presently there is no cure for the disease.
Molecular mechanism of HD pathogenesis
Increase in length of glutamine (Q) stretch at N-terminal of HTT, coded by exon1 of the gene, alters conformation of the protein leading to cytoplasmic and nuclear aggregates, also known as Neuronal Intra- Nuclear Inclusions (NII). It is still debatable whether monomer of conformation modified HTT, oligomer of the mutant protein or aggregates are toxic [6]. Aggregate/NII is observed in cell models, brains of transgenic animals and post-mortem brains of HD patients [7]. Aggregate formation is enhanced with increase in number of Q in vitro and in vivo and is believed to cause neurodegeneration [8]. Contradictory result that visible aggregates are protective to neurons is also available. Intermediate oligomers or modified monomer of mutant HTT has been proposed to be toxic [9,10]. Autosomal dominant nature of the disease suggests a toxic gain of function of mutated protein that disrupts normal cellular functions and causes neuronal death [11]. Loss of function of wild type protein may also contribute, at least partially, to disease pathology [12]. It is now believed that both loss of wild type HTT and toxic gain of function of mutant HTT result in HD pathogenesis. Alterations of various cellular processes like excitotoxicity, oxidative stress, endoplasmic reticulum stress, axonal transport, ubiquitin proteasome system, autophagy and apoptosis are implicated in HD. Besides mitochondrial dysfunction and transcriptional deregulation have been also implicated in HD and reviewed [13-15]. The summary of diverse molecular alterations observed in many studies leading to neuronal death is shown pictorially in Figure 1.
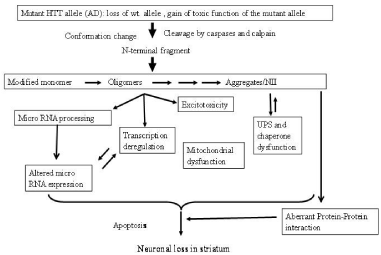
Figure 1: Molecular pathology of HD: various molecular events alter in HD.
Diverse approaches have been made to gather comprehensive global view of HD pathogenesis. These include identification of protein interacting partners of HTT, altered expression of protein coding genes or non-coding genes in HD using microarray or RNA sequencing in various models and post-mortem brains, modulators of HD pathogenesis in model systems and genome wide search for genetic markers that modulate AAO. To identify genes involve in HD pathogenesis, we collected data from the various sources. We also collected genes co-expressed with wild type HTT. Comparing the collected data, we made a comprehensive list of genes associated with HD. Enrichment analysis of HD associated genes for biological processes and pathways revealed that HD associated genes were over represented in processes and pathways already known to involve in HD pathogenesis.
Sources of Data and Methods
The flow chart for identification of HD associated genes and their possible involvement in HD pathogenesis and targets for treatment is shown in the Figure 2. Databases used to obtain various data related to HD associated genes, genetic variation in HD associated genes, HTT co expressed genes and others are shown in the Supplementary TEXT T1.
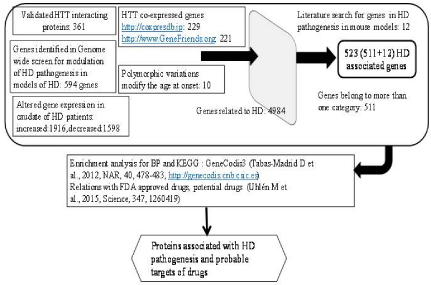
Figure 2: Flowchart for identification of HD associated genes, their
involvement in HD pathogenesis and possible drug targets. Numbers within
the box represent the number of genes identified in the category (see text
for detail).
HTT interacting proteins
To identifying function(s) of HTT, various interacting partners of HTT have been identified by Yeast 2 Hybrid (Y2H) assay [16- 18], affinity pull down followed by mass spectrometry assay and other large scale analysis [18-21]. Mutant HTT aggregate associated proteins were also identified by mass spectrometry [22]. Considering that Y2H predictions are subjected to high rate of false positives [23], it is necessary to establish physical interaction of the protein by a second independent method. In some cases, functional role of HTT interacting proteins were established [18]. We have considered validated HTT interacting protein if (a) interactions are confirmed by additional independent experiments, (b) interaction was observed in more than one large scale experiments and (c) HTT interacting proteins are shown to be functionally related to HD pathogenesis in models of HD. Several proteins have also been identified to interact with HTT and functionally associated with HD pathogenesis in models of HD in hypothesis based experiments [24-31]. We combined all data and catalogued 361 validated HTT interacting proteins. Details of results are shown in Supplementary TEXT T2 and the list of HTT interacting proteins is shown in Supplementary Table S1. Recently, about 100 interacting partners of HTT have been identified [32] and not included in our list. Sixteen proteins were common to our list of validated HTT interacting proteins.
Proteins identified in genome wide search for modulators of HD pathogenesis
Several small animal models of HD have been utilized to identify modulators of HD pathogenesis by using genome wide knocked down of genes and studying effects of mutant HTT for aggregates formation and/or toxicity. These models include S. cerevisiae (Yeast), worm (C. elegans), fly (D. melanogaster) and human/mouse cells in culture [33- 41]. Altogether 594 human homologous genes of yeast, fruitfly and C. elegans were identified in these studies that are likely to modulate the HD phenotypes. Detailed of result is shown in Supplementary Text T3 and genes are tabulated in Supplementary Table S2.
HTT co-expressed genes
Co-expressed genes provide important information of function (s) of genes and their possible common regulation. It has been shown that genes which are co-expressed are likely to be functionally related [42-46]. We have used two independent databases; Co-express db database (https://coxpresdb.jp) [45,46] and Gene Friends database (https://www.GeneFriends.org) [47] to identify genes co-expressed with wild type HTT. Genes co-expressed in at least two independent human and/or in another species with their chromosomal location of 229 HTT co-expressed genes are shown in the Supplementary Table S3, using Co-express db database. We collected 220 wild type HTT co-expressed protein coding genes from Gene Friends database (Supplementary Table S4) with correlation co-efficient = +0.445. Detailed processes for collecting these genes are described in Supplementary TEXT T4.
Altered expression of genes in caudate of HD patients
Data for altered expression of genes with p=0.001 was downloaded from published result [48]. In cases, more than one probe IDs for a particular gene, we have taken the maximum value of the relative expression. For down regulated genes, 3841 probe IDs and upregulated genes 3482 probe IDs were collected [48]. We have retained only those genes for which HGNC gene symbols and Entrez Gene IDs were available. Finally, we collected 1599 genes and 1914 gene that were significantly down regulated and upregulated respectively. This data is shown in Supplementary Tables S5A and S5B.
Polymorphic variations in protein coding genes associated with Age at Onset (AAO)
Age at Onset (AAO) defined as the time of appearance of first symptom(s) such as motor defects and cognitive impairment and has been shown to correlate inversely with number of CAG repeats in expanded allele of HTT gene. Investigations with large number of adult onset HD patients reveal that expanded CAG repeat number explains approximately 56% of the variations in AAO of motor sign. Additional genetic variations have also been shown to modify AAO and have been reviewed [49,50]. Genetic variations at putative promoters of protein coding gene neuropeptide Y (NPY) receptor NPY2R [5], HTT gene [51] and transcription factor E2F2 gene [52] have been identified to modify the AAO. These variations in promoters are shown to modify expression of the genes and involve in HD pathogenesis. Very recently, genome wide association studies identify various loci to be associated with AAO [53]. Copy- Number Variation (CNV) of SLC2A3 gene encodes neuronal glucose transporter GLUT3 could modulate AAO in HD. It is observed that increased dosage of SLC2A3 delayed AAO [54]. On the basis of these evidences and other experimental supports (Supplementary TEXT T5), we catalogued 12 genes namely OGG1, PPARGC1A, NPY, NRF- 1, SLC2A3/GLUT3, HTT, ATG7, CNR1, NPY2R, GRIN2A, HAP1 and ADORA2A are associated with HD.
Enrichment analysis for Biological Processes (BP) defined by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
To have an idea about functions of HD associated genes, we have used online resource of Genecodis3 (https://genecodis.dacya. ucm.es/). This facility can be used for enrichment analysis of various Gene Ontology (GO) descriptors for molecular functions, biological processes, cellular components and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. Proportion of genes in a particular GO term or KEGG pathway from input query list is computed and compared with those of proteins coded by human genome as catalogued in the database. Hypergeometric p value is computed after correction for multiple testing [55].
Drugs associated with the HD associated genes
Using known gene/protein targets of FDA approved small molecules, it has been shown that out of 625 enlisted protein targets in Drugbank, 516 proteins are targeted by small molecules, 83 proteins are targeted by biotech drugs and 26 proteins are targets of both small molecules and biotech drugs. These targets mainly belong to enzymes, transporters, voltage-gated ion channels; G-protein coupled receptors, nuclear receptors and CD markers. These proteins are also classified as Integral Membrane (IM), Single Pass Trans-Membrane (SPTM), IM/SPTM, secreted, membrane & secreted isoforms, and intracellular. In addition, 1054 genes classified as potential drug targets; which are not yet targets of FDA approved or experimental drugs in Drugbank database (www.drugbank.ca). These genes have categorized as probable targets based on similar properties of known FDA approved drug targets like protein families, structural folds, and biochemical similarities [56]. To find expression of the target genes in different 32 normal tissues, we observed that majority of target genes (~56%) were enhanced/enriched/group enriched (Supplementary TEXT T8) indicating genes expressed in restricted tissues are over represented among targets compared to housekeeping genes (~ 28% of drug targets are housekeeping genes (data not shown). We have utilized this data to identify if HD associated genes are also target of known drugs or potential drugs (https://www.proteinatlas.org/ humanproteome, accessed on June 1, 2016).
Result and Discussion
HD associated genes
We collected data from diverse sources to show that (a) out of more than 1000 interacting partners of HTT, 361 proteins interact with HTT in more than one experiment and/or functionally modify HD pathogenesis in model systems (HTT interacting), (b) 594 human homologue genes which were shown to modulate aggregate and/or toxicity induced by mutant HTT in genome wide knocked down studies in models of HD (Hdsi), (c) 230 genes including HTT co-expressed with wild type HTT in more than one species including human and/or in two independent human identified from Coexpress db, (d) 221 protein coding genes including HTT co-expressed with wild type HTT identified from Gene Friends database with correlation co-efficient = +0.445, (e) expression of 1598 genes that are decreased in caudate of HD patients with p=0.001 (Caudate down), (f) expression of 1914 genes that are increased in caudate of HD patients with p=0.001 (Caudate UP) and (g) genetic variations in 12 genes that modify AAO. Total number of unique genes is 4417 having 4930 relations indicating one gene is associated with more than one category. Number of protein related to more than one category was 523. We considered that these 523 proteins are associated with HD (HD associated protein). Detailed of choices of HD relations of genes in each category is described in the Supplementary TEXT T2-T6 and data is shown in Supplementary Tables S1-S5B, and summary of HD associated genes and their categories is shown in Supplementary Table S6. Among 523 HD associated genes, genetic variations in 3 genes modify AAO, expression of 212 gene was increased and 187 gene was decreased in caudate of HD patients, 91 and 76 genes were co-expressed with wild type HTT, 250 proteins interact with HTT, 195 proteins were coded by genes identified in genome wide study to modulate HD pathogenesis and denoted by Hdsi (Figure 3). Seven proteins were identified to be functionally related and observed/ identified in separate experiment to be in other categories. Five proteins were functionally related but not observed in any one of the categories and 48 proteins were functionally related and shown to interact with HTT. Diverse pathological conditions observed in HD may be due functional deregulation of these HD associated genes.
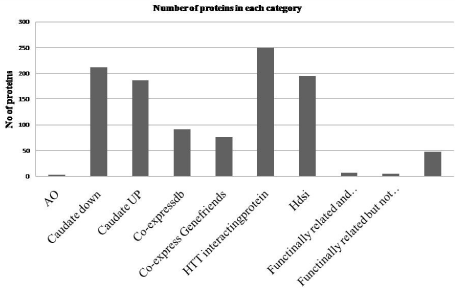
Figure 3: HD associated proteins in different category. In X-axis different
categories of proteins are shown; columns, 8, 9 and 10 represent functionally
related and observed/identified in separate experiment and categorized in
other categories, functionally related but not observed in any one of the
categories and proteins were functionally related and shown to interact with
HTT respectively (for detail please the text).
Enrichment of biological processes and KEGG pathways with HD associated genes
To find the biological processes defined by the Gene Ontology (GO) and the KEGG pathways involved with HD associated genes, we carried out enrichment analysis using GeneCodis3 (https:// genecodis.dacya.ucm.es/) (Section 2.6). Detailed result is shown in Supplementary Tables S7A and 7B and summarized in Table 1. Biological processes like apoptotic process (GO:0006915), signal transduction (GO:0007165), ubiquitin-dependent protein catabolic process (GO:0006511), positive regulation of transcription from RNA polymerase II promoter (GO:0045944), gene expression (GO:0010467), pathways like Huntington’s disease (Kegg:05016), protein processing in endoplasmic reticulum (Kegg:04141), regulation of actin cytoskeleton (Kegg:04810), oxidative phosphorylation (Kegg:00190), cell cycle (Kegg:04110) and apoptosis (Kegg:04210) were significantly enriched with HD associated proteins. Biological processes and pathways enriched were experimentally shown by many investigators to be involved in HD. In addition, enrichment of GO descriptor Cellular Component (CC) that indicates localization of proteins in organelles was also determined from GeneCodis3. Proteins are localized in different region/organelle (Supplementary Table S7C, Table 1).
![]()
Items
No. with p = 0.05
No with p =9.76E-05
Top 10 highly significantly enriched relevant descriptors
Unique protein in top 10 items
Biological Processes (BP)
611
86
nerve growth factor receptor signaling pathway (GO:0048011), apoptotic process (GO:0006915), signal transduction (GO:0007165), regulation of apoptotic process (GO:0042981), negative regulation of cell proliferation (GO:0008285), ubiquitin-dependent protein catabolic process (GO:0006511), synaptic transmission (GO:0007268), positive regulation of transcription from RNA polymerase II promoter (GO:0045944), gene expression (GO:0010467) and epidermal growth factor receptor signaling pathway (GO:0007173)
213 (323 relations)
KEGG pathways
114
49
Huntington's disease (Kegg:05016), Alzheimer's disease (Kegg:05010), Parkinson's disease (Kegg:05012), Neurotrophin signaling pathway (Kegg:04722), Focal adhesion (Kegg:04510), Protein processing in endoplasmic reticulum (Kegg:04141), Regulation of actin cytoskeleton (Kegg:04810), Oxidative phosphorylation (Kegg:00190), Cell cycle (Kegg:04110), Apoptosis (Kegg:04210)
120 (221 relations)
Cellular Component (CC)
210
49
cytoplasm (GO:0005737), cytosol (GO:0005829), nucleus (GO:0005634), nucleoplasm (GO:0005654), nucleolus (GO:0005730), protein complex (GO:0043234), plasma membrane (GO:0005886), mitochondrion (GO:0005739),cytoskeleton (GO:0005856) and perinuclear region of cytoplasm (GO:0048471)
442 (1120 relations)
Table 1: Summary of the enriched Biological processes and KEGG Pathways.
Among top enriched BPs, positive regulation of transcription from RNA polymerase II promoter (GO: 0045944) and gene expression (GO: 0010467) related to transcription regulation, are enriched with 63 unique HD associated gene (~12%). These genes include transcription regulators like TP53, CREB, HDAC1, HDAC2, HMGB1, HMGB2, KAT2B, LMO4, NFKB1, NFYA, SREBF2 and others, known to involve in HD pathogenesis [15,57-62]. Mediator complex subunits namely MED15 and MED23 interact with HTT are also in this group of protein. Mediator complex is known to regulate transcription [63]. Transcription associated BPs were also enriched with several ribosomal proteins (RPL13, RPL14, RPL15, RPL23, RPL6, RPL7 RPLP2 and RPS6). How ribosomal proteins regulate transcription remains unknown. RPL14 contains a “basic regionleucine zipper (bZIP)-like domain”; although its function remains unknown. RPL6 can activate transcription by binding specifically to domain C of the tax-responsive enhancer element of human T-cell leukemia virus type 1 [64]. RPS6 is component of the 40S ribosomal subunit and a substrate of protein kinases in ribosome. Recently, it has been shown that expression ribosomal S6 kinase gene RPS6KA1/ RSK1 is decreased in models of HD as well as in post-mortem brains of HD and compensation of the protein improves motor coordination, increased expression of synaptic markers and genes involved in synaptic plasticity [65].
Altogether 106 unique HD associated proteins were enriched with GO terms related to apoptosis (Supplementary Table S7A). KEGG pathway apoptosis (KEGG: 04210) was enriched with HD associated gene. Enhanced apoptosis is involved in HD and reviewed [66]. Targeting apoptosis is considered to be one of the approaches for treatment of neurodegenerative diseases in general and HD in particular [67]. Many GO terms related to cell cycle (Supplementary Table S7A) were enriched with 107 HD associated proteins. KEGG pathway cell cycle (KEGG: 04110) was also enriched significantly. Cell cycle abnormalities have been shown in different models of HD [68- 70]. Other major biological processes and pathways enriched with HD associated genes include signaling, proteasomal degradation, metabolism, differentiation and development, mRNA metabolic process including splicing, translation, vesicle-mediated transport and many others (Supplementary Table S7A and 7B). It is to be noted that one gene may be associated with more than one BP or pathway. These processes and pathways enriched with HD associated gene are already known to alter in HD or models of HD [71-86].
Pathways Huntington’s disease (KEGG: 05016), Alzheimer’s disease (KEGG: 05010) and Parkinson’s disease (KEGG: 05012) were the top 3 enriched (p= 1.95E-16) pathways with 48, 28 and 23 HD associated genes respectively. Altogether 54 unique HD associated genes are involved in these pathways; 16 proteins were common in 3 pathways. Common pathways among these diseases have been presented and common intervention approach have been proposed [87,88]. In summary, enrichment of diverse biological processes and pathways with HD associated genes collected in this manuscripts and known to be involved in HD show that these HD associated genes will not only be useful for deciphering molecular mechanisms of HD pathogenesis but also could be useful for targeting the processes/ pathways for intervention.
HD associated proteins: targets of FDA approved drugs and probable targets
To identify candidate of drug targets among HD associated proteins, we used protein targets of FDA approved drugs and probable drug targets as described in material and methods (section 2.7). Comparison of HD associated proteins with protein targets of drugs approved by FDA [56] revealed that 29 HD associated genes are targets of FDA approved drugs for other conditions. Similar comparison with proteins having similar characteristics of known drug targets, revealed that 52 HD associated proteins are probable drug targets (Table 2). Based on experimental evidences for HD modifying activity of proteins in various models of HD and other information including status of drug trial targeting a gene, a scoring system has been developed for probable targets of drugs for HD with Target Validation Score (TVS) by HD Research Crossroads as described [89]. Based on experimental evidence of involvement of proteins in HD pathogenesis and outcome of drug trials, TVS score varies between 0-5 (Supplementary TEXT T7) as described [89]. Maximum score of 5 was assigned to protein that demonstrated efficacy in a phase 3 clinical trial when drug or a gene therapy modulating the gene/protein (target) has been observed; minimum score of 0 was used when the gene is implicated in neurodegeneration or polyglutamine dysfunction based on genome-wide screens. TVS of HD related protein thus describes status of experimental validation of disease modifying effects of HD related proteins. Comparing proteins having TVS with HD associated genes described in the manuscript, we observed that 158 HD associated proteins had TVS; B4GALT6 only had TVS 0. It was evident that 136 proteins had TVS = 3; 17 proteins had TVS >3 [89] (Supplementary Tables 10A). Most of the HD associated genes described here was not catalogued [89] and thus did not have TVS. In large number of cases, expressions of genes were altered in caudate of HD and proteins coded by the genes are interacting partners of HTT (Supplementary Table S6). These genes could assign TVS with 1 according to HD Research Crossroads criteria. However, scoring system did not allow assigning a score to a protein, if a protein belongs to both categories. Similarly, if a gene was identified in genome wide screen, TVS is 0, however if the same gene belongs to another category like altered expression of genes in caudate of HD or HTT interacting partner, no scoring system is available. Thus there is scope for addition of scores to proteins, we described as HD associated proteins.
![]()
FDA approved drugs
ADORA2A /A2AR, ATP1A1, BCL2, CACNA2D1, CNR1, CPT2, DRD1, EGFR, FADS1, GRIN1, GRIN2A, GSS, HDAC1, HDAC2, HDAC3, HMGCR, KCNC1, MAPK1, MTOR, NFKB1, NR3C1, PAH, SLC25A4, SNAP25, TNFRSF8, PRKAA1/AMPKa1/AMPK, TUBB, TUBG1 and VAMP2 [29]
Probable targets of drugs
ADAR, ALDOA, APP, ASNS, ATM, ATP2A2, BAP1, CASK, CALM1, CASP10, CASP8, CTSD, CAV1, CHD8, CREBBP, CTH, CBS, CTNS, DLD, DNM1L, DNM2, ENTPD1, GPHN, GPI, HADHA, ITPR1, INPPL1, LDHA, NDUFS1, NDUFV1, NUP98, PPIB, PFKM, PGM1, PTK2B, POMT2, SNRNP200, SLC16A1, SLC25A12, SLC25A13, SLC25A3, SPG7, SUCLA2, SYP, STX1A, SNCA, TMPRSS5, TP53, TYK2, UQCRC2, AKT1, WHSC1 [52]
Proteins related to HD having TVS >3
GRIN1, ADORA2A /A2AR, HDAC1, HTT, ITPR1, MAP3K10, ATM, CALM1, CASP1, CAV1, DLD, HSPA4, SIRT1, SP1, TGM2, TP53, ZDHHC17 [17]
Table 2: HD associated proteins and their status for drug targets.
Novel targets: not included in HD Research Crossroads list with TVS
We identified 81 HD associated proteins (Table 3) which are either targets of FDA approved drugs or probable target. Among these proteins, TVS was assigned to 32 proteins and TVS was not assigned to 49 proteins. Among 32 proteins, 15 proteins are FDA approved drug targets; 17 proteins are probable targets. Among 49 proteins without TVS, 14 proteins are FDA approved drug targets and 35 proteins are probable drug targets (Table 3, Supplementary Tables S8A-S8F). These 49 proteins are likely to be novel targets for drugs. Further characterization is necessary to validate the proposition.
![]()
Category
HD associated proteins (no)
With Target Validation Score (TVS)
Without TVS
Target of FDA approved drugs
ADORA2A/A2AR, ATP1A1, BCL2, CNR1, DRD1, GRIN1, GRIN2A, HDAC1, HDAC2, HDAC3, MAPK1, NFKB1, SLC25A4, SNAP25 and TUBB [15]
CACNA2D1, CPT2, EGFR, FADS1, GSS, HMGCR, KCNC1, MTOR, NR3C1, PAH, TNFRSF8, PRKAA1/
AMPKa1/AMPK, TUBG1 and VAMP2 [14]
Probable targets
AKT1, ATM, CALM1, CASP8, CAV1, CBS, CREBBP, CTSD, DLD, DNM1L, DNM2, GPI, ITPR1, NDUFV1, SNRNP200, STX1A, TP53 [17]
ADAR, ALDOA, APP, ASNS, ATP2A2, BAP1, CASK, CASP10, CHD8, CTH, CTNS, ENTPD1, GPHN, HADHA, INPPL1, LDHA, NDUFS1, NUP98, PFKM, PGM1, POMT2, PPIB, PTK2B, SLC16A1, SLC25A12, SLC25A13, SLC25A3, SNCA, SPG7, SUCLA2, SYP, TMPRSS5, TYK2, UQCRC2, WHSC1 [35]
Table 3: HD associated proteins which are FDA approved drug targets or probable drug targets.
Out of 15 HD associated proteins, which are targets of FDA approved drugs and were assigned with TVS, expression of 5 proteins namely CNR1, DRD1, GRIN1, GRIN2A and SNAP25 were enriched/ enhanced in brains; expression of SLC25A4 (group enriched in heart muscle, skeletal muscle) and ADORA2A /A2AR (tissue enhanced in heart muscle, skeletal muscle) enhanced in a group of tissues other than brain. Other 8 genes (ATP1A1, HDAC1, HDAC2, HDAC3, BCL2, NFKB1, MAPK1 and TUBB) expressed almost equally in 32 tissues and likely to be housekeeping genes. Biological processes and pathways associated with these genes are shown in the Supplementary Table S9A. Activation of DRD1 gene and ADORA2A /A2AR is known to increase cAMP-dependent Protein Kinase (PKA) through phosphorylation its substrates and impairs cognition in mouse model of HD [90]. Reduced expression of DRD1 and ADORA2A/A2AR in caudate of HD patients (Supplementary Table S5A) was observed in many other studies. Apparent contradiction of decreased expression and downstream PKA activation was explained by altered density and function of both DRD1 and ADORA2A/A2AR in hippocampus of R6/1 mice. Combined inactivation of DRD1 and ADORA2A /A2AR with agonists protects effects of mutant HTT [91]. DRD1 is associated with 55 biological processes like dopamine transport (GO: 0015872), long term synaptic depression (GO: 0060292), glucose import (GO: 0046323) and 10 KEGG pathways like cAMP signaling pathway (KEGG: 04024), neuroactive ligand-receptor interaction (KEGG: 04080), Parkinson’s disease (KEGG: 05012) could be relevant for HD. Among 56 BPs associated with ADORA2A /A2AR, GABAergic (GO: 0032230), many apoptosis related GO terms, regulation of synaptic plasticity (GO:0048167), regulation of mitochondrial membrane potential (GO:0051881), regulation of excitatory postsynaptic membrane potential (GO:0060079) may have significance for HD. Pathways like calcium signaling pathway (KEGG: 04020), cAMP signaling pathway, Neuroactive ligand-receptor interaction and Parkinson’s disease pathway may be associated with HD, as discussed in section 3.2. Activation of the DRD1 and ADORA2A/A2AR by pharmacological inhibitors is likely to change these processes and pathways and beneficial for HD.
GRIN1 had the highest TVS of 4.5; modulation of the gene demonstrated efficacy in a phase 2 clinical trial [89]. Our analysis showed that GRIN1 was identified in genome wide study to suppress effect of mutant HTT [41] and expression of GRIN1 was decreased in caudate of HD (Supplementary Table S5A). Among 32 normal tissues, expression of GRIN1 in normal brain was maximum compared to other tissues [56]. GRIN1 is associated 54 BPs like positive regulation of transcription from RNA polymerase II promoter (GO: 0045944), regulation of excitatory postsynaptic membrane potential (GO: 0060079), ionotropic glutamate receptor signaling pathway (GO: 0035235), calcium ion homeostasis (GO: 0055074) and 15 pathways like Huntington’s disease (Kegg:05016), Alzheimer’s disease (Kegg:05010), Calcium signaling pathway (Kegg:04020), Amyotrophic lateral sclerosis (Kegg:05014), Long-term potentiation (Kegg:04720) (Supplementary Table S9A). Genetic variation in Cannabinoid Receptor 1 (CNR1) modifies AAO [92]. Expression of CNR1was decreased in HD brain [48], pharmacological inhibitors increased effects of mutant HTT in mice model [93] and HD mouse with CNR1+ background showed recovery of loss of excitatory striatal synapses [94]. CNR1 is associated with 36 BPs like positive regulation of apoptotic process (GO: 0043065) and 3 pathways namely retrograde endocannabinoid signaling (KEGG: 04723), neuroactive ligandreceptor interaction (KEGG: 04080) and Rap1 signaling pathway (KEGG: 04015), known to involve in HD. Activation of CNR1 could thus be beneficial for HD. SNAP25, a synaptosomal-associated protein, interacts with HTT, over expression SNAP25 rescues effects of mutant HTT [18,40], expression was decreased in caudate of HD brain [48,95], belongs to synaptic vesicle cycle (KEGG: 04721) and insulin secretion (KEGG: 04911) pathways (Supplementary Table S9A). All 5 genes are expressed significantly higher level in normal brain compared to other tissues. On the basis of expression of genes in normal brain and being target of FDA approved drugs and involved in diverse HD relevant biological processes and pathways, we provided additional support that HD associated protein CNR1, DRD1, GRIN1, GRIN2A and SNAP25 having different TVS could be possible drug targets.
Characterization of 49 proteins without TVS score: new targets for treatment of HD
Supplementary Table S8FF shows 49 proteins; out of which 14 proteins are targets of FDA approved drugs and 35 proteins are potential drug target. These genes do not have TVS. Expression of KCNC1, SNCA, SYP and TMPRSS5 were enriched /enhanced in a group of normal tissues including brain. Expressions of EGFR, PAH, CTH, NUP98 were enhanced/enriched in normal placenta, (kidney and liver), liver and (heart and skeletal muscle) respectively. KCNC1 and TMPRSS5 are expressed in normal brain, SNCA is expressed in bone marrow and brain and SYP is expressed in brain and salivary gland at significantly higher level compared to other normal tissues. All other 41 genes expressed ubiquitously in all 32 tissues (Supplementary Table S8F). Biological processes and pathways associated with these genes are shown in Supplementary Table S9B. Role of KCNC1 and TMPRSS5 in HD remains unknown. Both the proteins were identified in genome wide screen; KCNC1 enhances the effects of mutant HTT in model of HD [40], while TMPRSS5 protects the effects of mutant HTT [41]. Expression of KCNC1 was decreased and expression of TMPRSS5 was increased in caudate of HD patients [48]. KCNC1 is associated with biological process ion transport (GO: 0006811), potassium ion transport (GO: 0006813), synaptic transmission (GO: 0007268), transmembrane transport (GO: 0055085) and protein homooligomerization (GO: 0051260), former 4 processes are known to alter in HD. TMPRSS5 is associated with BP receptor-mediated endocytosis (GO: 0006898) and localized in neuronal cell body (GO: 0043025) indicating possible role in neuronal functions. SYP (synaptophysin) interacts with HTT [18,21], expression was decreased in brain [48,96], associated with 7 BPs [endocytosis (GO:0006897), synaptic vesicle maturation (GO:0016188), regulation of long-term neuronal synaptic plasticity (GO:0048169), regulation of shortterm neuronal synaptic plasticity (GO:0048172), synaptic vesicle membrane organization (GO:0048499), cellular response to organic substance (GO:0071310), regulation of opioid receptor signaling pathway (GO:2000474)]. Aggregate of SNCA was observed in HD, independent of aggregate of mutant HTT [97]. Exogenous expression SNCA or deletion of SNCA increased or decreased respectively the pathogenesis in mouse models of HD [98]. SNCA is associated with 68 BPs; among them HD related processes are dopamine metabolic process (GO:0042417), mitochondrial ATP synthesis coupled electron transport (GO:0042775), negative regulation of apoptotic process (GO:0043066), negative regulation of neuron apoptotic process (GO:0043524), positive regulation of endocytosis (GO:0045807), negative regulation of exocytosis (GO:0045920), regulation of neuronal synaptic plasticity (GO:0048168), synaptic vesicle endocytosis (GO:0048488), synaptic vesicle transport (GO:0048489), positive regulation of synaptic transmission (GO:0050806), synapse organization (GO:0050808) and oxidationreduction process (GO:0055114). SNCA is also associated with Parkinson’s disease (KEGG: 05012) and Alzheimer’s disease (KEGG: 05010) pathways. Thus drug inactivating SNCA would likely to be an effective treatment for HD.
In summary, KCNC1, a target of FDA approved drug, SNCA, SYP and TMPRSS5, probable targets of drugs could be new targets for HD. Three proteins, except SNCA, are not well studied for their role in HD and require further investigations. The result is summarized in Figure 4.
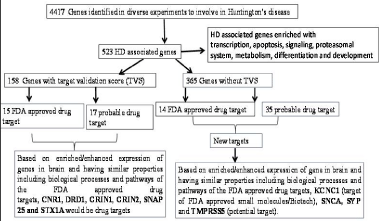
Figure 4: Graphical summary of the result.
Other targets
Based on the targets of FDA approved or probable drug targets and enhanced/enriched expression of genes in normal brain compared to other tissues among HD associated genes, we proposed additional probable drug targets. However several other genes are also shown to be the target of drugs. Tetrabenazine, an inhibitor of monoamine transporter SLC18A2/VMAT2, has been approved by FDA for treatment of chorea [99]. Inhibitions of MAOA, MAOB, HDAC1, HDAC2, HDAC3 and PRKAA1 (AMPK1/ AMPKa1) protect pathological effects in models of HD. Allele specific silencing of mutant HTT reduces aggregates and other effects of mutant HTT. These genes are associated with biological processes and pathways relevant for HD (Supplementary TEXT T9).
Conclusion
Comprehensive literature and database search identified 523 genes designated as HD associated genes. Enrichment analysis revealed that HD associated genes were significantly over represented in biological processes and pathways known to be involved in HD pathogenesis. These HD associated genes may be used for understating defects in molecular abnormalities in HD. Comparing HD associated proteins with proteins catalogued as probable targets in HD Research Crossroads with target validation scores, targets of FD approved drugs and probable targets, we identified 49 probable new targets. We emphasized that KCNC1, a target of FDA approved small molecules/Biotech, SYP and TMPRSS5, probable drug target, expressions enriched/enhanced in brains and predicted membrane protein, secreted/transporter protein would be new targets of drugs for treatment of HD. Based on expression of genes enhanced/enriched in normal brain, we further showed that genes SNCA, CNR1, DRD1, GRIN1, GRIN2A and SNAP25 with different TVS could be important targets. Except SNCA, all five genes are targets of FDA approved drugs. HD associated genes collated here will be useful to understand the molecular pathology in HD as well as protein/biological process/ pathway targets for treatment of HD.
References
- Huntington G. On Chorea. The Medical and Surgical Reporter of Philadelphia: A Weekly Journal. 1872; 26: 317-321.
- Cardoso F, Seppi K, Mair KJ, Wenning GK, Poewe W. Seminar on choreas. Lancet Neurol. 2006; 5: 589-602.
- Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010; 5: 40.
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993; 72: 971-983.
- van der Burg JM, Björkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009; 8: 765-774.
- Takahashi T, Katada S, Onodera O. Polyglutamine diseases: where does toxicity come from? what is toxicity? where are we going? J Mol Cell Biol. 2010; 2: 180-191.
- Hoffner G, Souès S, Djian P. Aggregation of Expanded Huntingtin in the Brains of Patients with Huntington Disease. Prion. 2007; 1: 26-31.
- Ross CA. Poirier Protein aggregation and neurodegenerative disease. Nat Med. 2004; 10 Suppl: S10-S17.
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004; 431: 805-810.
- Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, et al. Identification of Novel Potentially Toxic Oligomers Formed in Vitro from Mammalian-derived Expanded huntingtin Exon-1 Protein. The J Biol Chem. 2012; 287: 16017-16028.
- Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington's disease. Curr Top Dev Biol. 2006; 75: 25-71.
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001; 24: 182-188.
- Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, et al. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008; 412: 191-209.
- Ross CA. Tabrizi, SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011; 10: 83-98.
- Seredenina T, Luthi-Carter R. What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis. 2011; 45: 83-98.
- Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet. 1998; 7: 1463-1474.
- Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U, et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington's disease. Mol Cell. 2004; 15: 853-865.
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007; 3: e82.
- Culver BP, Savas JN, Park SK, Choi JH, Zheng S, Zeitlin SO, et al. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem. 2012; 287: 21599-21614.
- Ratovitski T, Chighladze E, Arbez N, Boronina T, Herbrich S, Cole RN, et al. Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis. Cell Cycle. 2012; 11: 2006-2021.
- Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012; 75: 41-57.
- Mitsui K, Nakayama H, Akagi T, Nekooki M, Ohtawa K, Takio K, et al. Purification of polyglutamine aggregates and identification of elongation factor1alpha and heat shock protein 84 as aggregate-interacting proteins. J Neurosci. 2002; 22: 9267-9277.
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002; 417: 399-403.
- McFarland KN, Huizenga MN, Darnel SB, Sangrey GR, Berezovska O, Cha JH, et al. MeCP2: a novel Huntingtin interactor. Hum Mol Genet. 2014; 23: 1036-1044.
- Sanders SS, Mui KK, Sutton LM, Hayden MR. Identification of binding sites in Huntingtin for the Huntingtin Interacting Proteins HIP14 and HIP14L. PLoS One. 2014; 9: e90669.
- Tourette C, Li B, Bell R, O'Hare S, Kaltenbach LS, Mooney SD, et al. A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J Biol Chem. 2014; 289: 6709-6726.
- Trushina E, Canaria CA, Lee DY, McMurray CT. Loss of caveolin-1 expression in knock-in mouse model of Huntington's disease suppresses pathophysiology in vivo. Hum Mol Genet. 2014; 23: 129-144.
- Rutherford NJ, Lewis J, Clippinger AK, Thomas MA, Adamson J, Cruz PE, et al. Unbiased screen reveals ubiquilin-1 and -2 highly associated with huntingtin inclusions. Brain Res. 2013; 1524: 62-73.
- Sutton LM, Sanders SS, Butland SL, Singaraja RR, Franciosi S, Southwell AL, et al. Hip14l-deficient mice develop neuropathological and behavioural features of Huntington disease. Hum Mol Genet. 2013; 22: 452-465.
- Roux JC, Zala D, Panayotis N, Borges-Correia A, Saudou F, Villard L. Modification of Mecp2 dosage alters axonal transport through the Huntingtin/Hap1 pathway. Neurobiol Dis. 2012; 45: 786-795.
- Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, et al. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002; 11: 2815-2828.
- Hosp F, Vossfeldt H, Heinig M, Vasiljevic D, Arumughan A, Wyler E, et al. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 2015; 11: 1134-1146.
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ. Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science. 2003; 302: 1769-1772.
- Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, et al. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation, Proc Natl Acad Sci USA. 2004; 101: 6403-6408.
- Branco J, Al-Ramahi I, Ukani L, Pérez AM, Fernandez-Funez P, Rincón-Limas D, et al. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum Mol Genet. 2008; 17: 376-390.
- Doumanis J, Wada K, Kino Y, Moore AW, Nukina N. RNAi screening in Drosophila cells identifies new modifiers of mutant huntingtin aggregation. PLoS One. 2009; 4: e7275.
- Zhang S, Binari R, Zhou R, Perrimon N. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics. 2010; 184: 1165-1179.
- Teuling E, Bourgonje A, Veenje S, Thijssen K, de Boer J, van der Velde J, et al. Modifiers of mutant huntingtin aggregation: functional conservation of C. elegans-modifiers of polyglutamine aggregation. PLoS Curr. 2011; 3: RRN1255.
- Chen X, Burgoyne RD. Identification of common genetic modifiers of neurodegenerative diseases from an integrative analysis of diverse genetic screens in model organisms. BMC Genomics. 2012; 13: 71.
- Lejeune FX, Mesrob L, Parmentier F, Bicep C, Vazquez-Manrique RP, Parker JA, et al. Large-scale functional RNAi screen in C. elegans identifies genes that regulate the dysfunction of mutant polyglutamine neurons. BMC Genomics. 2012; 13: 91.
- Miller JP, Yates BE, Al-Ramahi I, Berman AE, Sanhueza M, Kim E, et al. A genome-scale RNA-interference screen identifies RRAS signaling as a pathologic feature of Huntington's disease. PLoS Genet. 2012; 8: e1003042.
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998; 95: 14863-14868.
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003; 302: 249-255.
- Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc Natl Acad Sci USA. 2006; 103:17973-17978.
- Obayashi T, Hayashi S, Shibaoka M, Saeki M, Ohta H, Kinoshita K. COXPRESdb: a database of coexpressed gene networks in mammals. Nucleic Acids Res. 2008; 36(Database issue): D77-D82.
- Obayashi T, Okamura Y, Ito S, Tadaka S, Motoike IN, Kinoshita K. COXPRESdb: a database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 2013; 41(Database issue): D1014-D1020.
- van Dam S, Craig T, de Magalhes JP. GeneFriends: a human RNA-seq-based gene and transcript co-expression database. Nucleic Acids Res. 2015; 43(Database issue): D1124-D1132.
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006; 15: 965-977.
- Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington's disease. Mov Disord. 2014; 29: 1359-1365.
- Arning L, Epplen JT. Genetic modifiers in Huntington's disease: fiction or fact? Neurogenetics. 2013; 14: 171-172.
- Beanović K, Nørremølle A, Neal SJ, Kay C, Collins JA, Arenillas D, et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci. 2015; 18: 807-816.
- Valcárcel-Ocete L, Alkorta-Aranburu G, Iriondo M, Fullaondo A, García-Barcina M, Fernández-García JM, et al. Exploring Genetic Factors Involved in Huntington Disease Age of Onset: E2F2 as a New Potential Modifier Gene. PLoS One. 2015; 10: e0131573.
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell. 2015; 162: 516-526.
- Vittori A, Breda C, Repici M, Orth M, Roos RA, Outeiro TF, et al. Copy-number variation of the neuronal glucose transporter gene SLC2A3 and age of onset in Huntington's disease. Hum Mol Genet. 2014; 23: 3129-3137.
- Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012; 40: W478-483.
- Uhlén M, Fagerberg Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, et al. Tissue-based map of the human proteome. Science. 2015; 347: 1260419.
- Diamanti D, Mori E, Incarnato D, Malusa F, Fondelli C, Magnoni L, et al. Whole gene expression profile in blood reveals multiple pathways deregulation in R6/2 mouse model. Biomark Res. 2013; 1: 28.
- Valor LM, Guiretti D, Lopez-Atalaya JP, Barco A. Genomic landscape of transcriptional and epigenetic dysregulation in early onset polyglutamine disease. J Neurosci. 2013; 33: 10471-10482.
- Kumar A, Vaish M, Ratan RR. Transcriptional dysregulation in Huntington's disease: a failure of adaptive transcriptional homeostasis. Drug Discov Today. 2014; 19: 956-962.
- Durrenberger PF, Fernando FS, Kashefi SN, Bonnert TP, Seilhean D, Nait-Oumesmar B, et al. Common mechanisms in neurodegeneration and neuroinflammation: a Brain Net Europe gene expression microarray study. J Neural Transm (Vienna). 2015; 122: 1055-1068.
- Kerschbamer E, Biagioli M. Huntington's Disease as Neurodevelopmental Disorder: Altered Chromatin Regulation, Coding, and Non-Coding RNA Transcription. Front Neurosci. 2016; 9: 509.
- Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016; 39: 221-234.
- Poss ZC, Ebmeier CC, Taatjes DJ. The Mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013; 48: 575-608.
- Morita T, Sato T, Nyunoya H, Tsujimoto A, Takahara J, Irino S, et al. Isolation of a cDNA clone encoding DNA-binding protein (TAXREB107) that binds specifically to domain C of the tax-responsive enhancer element in the long terminal repeat of human T-cell leukemia virus type I. AIDS Res Hum Retroviruses. 1993; 9: 115-121.
- Anglada-Huguet M, Giralt A, Rué L, Alberch J, Xifró X. Increased 90-kDa ribosomal S6 kinase (Rsk) activity is protective against mutant huntingtin toxicity. Biochim Biophys Acta. 2016; 1862: 1255-1266.
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014; 112: 24-49.
- Brett AC, Rosenstock TR, Rego AC. Current therapeutic advances in patients and experimental models of Huntington's disease. Curr Drug Targets. 2014; 15; 313-334.
- Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, et al. Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington's disease. Hum Mol Genet. 2015; 24: 1602-1616.
- Das E, Jana NR, Bhattacharyya NP. Delayed Cell Cycle Progression in STHdh(Q111)/Hdh(Q111) Cells, a Cell Model for Huntington's Disease Mediated by microRNA-19a, microRNA-146a and microRNA-432. Microrna. 2015; 4: 86-100.
- Molina-Calavita M, Barnat M, Elias S, Aparicio E, Piel M, Humbert S. Mutant huntingtin affects cortical progenitor cell division and development of the mouse neocortex. J Neurosci. 2014; 34: 10034-10040.
- Bowles KR, Brooks SP, Dunnett SB, Jones L. Huntingtin Subcellular Localisation Is Regulated by Kinase Signalling Activity in the StHdhQ111 Model of HD. PLoS One. 2015; 10: e0144864.
- Naia L, Ferreira IL, Cunha-Oliveira T, Duarte AI, Ribeiro M, Rosenstock TR, et al. Activation of IGF-1 and Insulin Signaling Pathways Ameliorate Mitochondrial Function and Energy Metabolism in Huntington's Disease Human Lymphoblasts. Mol Mol Neurobiol. 2015; 51: 331-348.
- Sepers MD, Raymond LA. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington's disease. Drug Discov Today. 2014; 19: 990-996.
- Nambron R, Silajdić E, Kalliolia E, Ottolenghi C, Hindmarsh P, Hill NR, et al. A Metabolic Study of Huntington's Disease. PLoS One. 2016; 11: e0146480.
- Handley RR, Reid SJ, Patassini S, Rudiger SR, Obolonkin V, McLaughlan CJ, et al. Metabolic disruption identified in the Huntington's disease transgenic sheep model. Sci Rep. 2016; 6: 20681.
- Chen JY, Tran C, Hwang L, Deng G, Jung ME, Faull KF, et al. Partial Amelioration of Peripheral and Central Symptoms of Huntington's Disease via Modulation of Lipid Metabolism. J Huntingtons Dis. 2016; 5: 65-81.
- Karasinska JM, Hayden MR. Cholesterol metabolism in Huntington disease. Nat Rev Neurol. 2011; 7: 561-572.
- Wade BE, Wang CE, Yan S, Bhat K, Huang B, Li S, et al. Ubiquitin-activating enzyme activity contributes to differential accumulation of mutant huntingtin in brain and peripheral tissues. J Neurosci. 2014; 34: 8411-8422.
- Molero AE, Arteaga-Bracho EE, Chen CH, Gulinello M, Winchester ML, Pichamoorthy N, et al. Selective expression of mutant huntingtin during development recapitulates characteristic features of Huntington's disease. Proc Natl Acad Sci USA. 2016; 113: 5736-5741.
- Humbert S. Is Huntington disease a developmental disorder? EMBO Rep. 2010; 11: 899.
- Cabrera JR, Lucas JJ. MAP2 splicing is altered in Huntington's disease. Brain Pathol. 2016.
- Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA. 2013; 110: 2366-2370.
- White JA, Banerjee R, Gunawardena S. Axonal Transport and Neurodegeneration: How Marine Drugs Can Be Used for the Development of Therapeutics. Mar Drugs. 2016; 14: E102.
- Steinert JR, Campesan S, Richards P, Kyriacou CP, Forsythe ID, Giorgini F. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington's disease. Hum Mol Genet. 2012; 2: 2912-2922.
- Lee J, Hwang YJ, Ryu H, Kowall NW, Ryu H. Nucleolar dysfunction in Huntington's disease. Biochim Biophys Acta. 2014; 1842: 785-790.
- Tauber E, Miller-Fleming L, Mason RP, Kwan W, Clapp J, Butler NJ, et al. Functional gene expression profiling in yeast implicates translational dysfunction in mutant huntingtin toxicity. J Biol Chem. 2011; 286: 410-419.
- Richards RI, Robertson SA, O'Keefe LV, Fornarino D, Scott A, Lardelli M, et al. The Enemy within: Innate Surveillance-Mediated Cell Death, the Common Mechanism of Neurodegenerative Disease. Front Neurosci. 2016; 10: 193.
- Fan HC, Chi CS, Cheng SN, Lee HF, Tsai JD, Lin SZ, et al. Targeting New Candidate Genes by Small Molecules Approaching Neurodegenerative Diseases. Int J Mol Sci. 2015; 17: E26.
- Kalathur RK, Hernández-Prieto MA, Futschik ME. Huntington's disease and its therapeutic target genes: a global functional profile based on the HD Research Crossroads database. BMC Neurol. 2012; 12: 47.
- Giralt A, Saavedra A, Carretón O, Xifró X, Alberch J, Pérez-Navarro E. Increased PKA signaling disrupts recognition memory and spatial memory: role in Huntington's disease. Hum Mol Genet. 2011; 20: 4232-4247.
- Tyebji S, Saavedra A, Canas PM, Pliassova A, Delgado-García JM, Alberch J, et al. Hyperactivation of D1 and A2A receptors contributes to cognitive dysfunction in Huntington's disease. Neurobiol Dis. 2015; 74: 41-57.
- Kloster E, Saft C, Epplen JT, Arning L. CNR1 variation is associated with the age at onset in Huntington disease. Eur J Med Genet. 2013; 56: 416-419.
- Blázquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, Resel E, et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain. 2011; 134(Pt 1): 119-136.
- Naydenov AV, Sepers MD, Swinney K, Raymond LA, Palmiter RD, Stella N. Genetic rescue of CB1 receptors on medium spiny neurons prevents loss of excitatory striatal synapses but not motor impairment in HD mice. Neurobiol Dis. 2014; 71: 140-150.
- Smith R, Klein P, Koc-Schmitz Y, Waldvogel HJ, Faull RL, Brundin P, et al. Loss of SNAP-25 and rabphilin 3a in sensory-motor cortex in Huntington's disease. J Neurochem. 2007; 103:115-123.
- Goto S, Hirano A. Synaptophysin expression in the striatum in Huntington's disease. Acta Neuropathol. 1990; 80: 88-91.
- Tomás-Zapico C, Díez-Zaera M, Ferrer I, Gómez-Ramos P, Morán MA, Miras-Portugal MT, et al. a-Synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington's disease. Hum Mol Genet. 2012; 21: 495-510.
- Corrochano S, Renna M, Carter S, Chrobot N, Kent R, Stewart M, et al. a-Synuclein levels modulate Huntington's disease in mice. Hum Mol Genet. 2012; 21: 485-494.
- Frank S. Tetrabenazine: the first approved drug for the treatment of chorea in US patients with Huntington disease. Neuropsychiatr Dis Treat. 2010; 6: 657-665.