
Case Series
Austin J Genet Genomic Res. 2024; 5(1): 1024.
Cytogenetic Evaluation of Cleft Lip and/or Palate
Osman Demirhan*
Department of Medical Biology and Genetics, Faculty of Medicine, Çukurova University, Balcali- Adana/Turkey
*Corresponding author: Osman Demirhan, Prof. PhD Çukurova University, Faculty of Medicine, Department of Medical Biology and Genetics, Adana-Turkey. Email: odemirhan42@gmail.com; osdemir@cu.edu.tr
Received: December 19, 2023 Accepted: January 17, 2024 Published: January 23, 2024
Abstract
Purpose: It is known that the possible causes of congenital defects such as common cleft palate and/or libidinal (CLP) are multifactorial and occur as a result of genetic and environmental risk factors. Although these defects appear to have a genetic cause, the cause of most cases is still unknown. In this study, the relationship of CLP to possible cytogenetic causes was evaluated. In this study, the relationship of CLP with possible cytogenetic causes was evaluated.
Material and Methods: In this study, conventional karyotyping was performed on 10 patients who were referred to our genetics laboratory with complaints of cleft lip and/or palate.
Results: Structural and numerical CAs were found in four of the 10 patients with CLP, and a normal karyotype was found in 6 of them. Two male patients had XXY karyotype, one had 22q12 trisomy and one had pericentric inversion on chromosome 9.
Conclusion: Our findings support that X chromosome abundance and dosage of some genes in the 22q12 region may affect the development of cleft palate and lip and contribute to CLP. However, it has provided new opportunities for the understanding of orofacial cleft (OFC) biology and clinical research and has demonstrated the need to provide medical-genetic counseling to parents of children with gene and chromosomal disorders.
Keywords: Cleft lip and palate; Etiology; Sitogenetics; XXY karyotype; 22q12 trisomy
Introduction
CLP is the most common and best-known congenital orofacial birth defect in humans. There has recently been evidence to support a multifactorial inheritance pattern. In a large series of patients, some appear to result from single mutant genes, some from chromosomal abnormalities, some from specific environmental agents, and some from the interaction of many genetic and environmental differences, each with a relatively minor effect [1]. The average incidence for CL/P is reported to be around 1 in 700 newborns [2,3]. A cleft palate is seen in 1 in 1500 newborn babies. The incidence of cleft lip and palate in Turkey has been reported as 1.1 per thousand, and the incidence of isolated cleft palate as 0.77 per thousand [4]. Although these defects seem more likely due to a genetic cause, the cause of most cases is still unknown. These defects may occur alone or as part of a wide variety of chromosomal, gene, or teratogenic syndromes. Although significant progress has been made in identifying genetic and environmental triggers for syndromic ones, the etiology of non-syndromic forms has not yet been fully defined. Because of the heterogeneous etiology of CLP, it is necessary to know the biology of facial development and how environmental risks interact with genetic factors. Genome-wide linkage and association studies have identified new loci with significant relevance. There are significant phenotypic differences in individuals and family members with orofacial cleft birth defects, and the incidence of structural and numerical chromosomal changes in patients has been reported as 3.6% [5]. This study may provide information to better interrogate cytogenetic analysis for loci other than CLP-related coding regions. Our knowledge on this topic is largely biased, so more extensive research is needed to understand the mechanisms underlying these defects.
Methodology
Methods
Ten children from the pediatric clinic with complaints of cleft palate and lip were sent to our laboratory for genetic analysis. The male/female ratio of the cases was 4/6 (0.7), the mean gestational week was 37.7, and the mean maternal age was 26.0. These children ranged from 17 days to 3 years (mean 1.2 years). There was only cleft palate in one of the cases, and both cleft palate and lip in the others. A seventeen-day-old boy with cleft lip and cleft palate was admitted to the neonatal service because he could not be fed. The patient's parents were the first children of a 25-year-old mother and a 27-year-old father, who were second-degree relatives. There was no similar disease in her family history, and her physical examination revealed a low ear, hypertelorism, bilateral cleft lip, and complete cleft palate deforming the nasal root, low hairline, and pes equinovarus (Figure 1,2). A three-year-old boy was sent for genetic analysis for unilateral cryptoorchidism. The patient had previously been operated for cleft palate and lip, and there were surgical scars on the upper lip and philtrum (Figure 3,4). The patient's parents were related, he was the first child of a 23-year-old healthy mother and 29-year-old healthy father, and there was a diagnosis of CLP in his family history. Other children were those with only cleft palate or cleft lip and palate defect without any other congenital defect. There was consanguineous marriage between the parents of four cases. Conventional karyotyping was performed using 4 ml of peripheral blood for sowing, harvesting, banding, and analysis. All procedures performed in studies were in accordance with the ethical standards of the institutional and/or national research committee (Scientific Research Ethics Committee Directive) and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
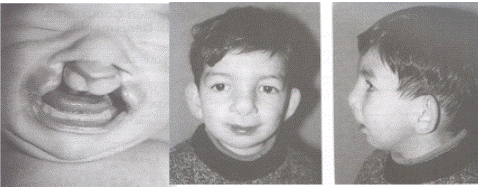
Figure 1: The facial appearance of two cases with 47, XXY karyotype.
Results
Numerical and structural damage was found in 4 of 10 patients analyzed. Two of these 4 patients had 47, XXY, one 47, XX, del(22q12 > qter) or partial trisomy 22q12, and one had inv(9)(p11;q13). The other 6 cases had normal chromosome establishment.
Discussion
Genetic abnormalities are known predisposing factors for syndromic and non-syndromic OFC, and many genes and loci involved in its etiology have been identified. Nevertheless, no consensus has been reached regarding inheritance patterns. Many genetic and environmental factors play a role in most clefts, as well as polygenic factors. Genetic factors cause clefts in about 20% to 50% of cases, while the rest can be attributed to environmental factors or gene-environment interactions. In the present study, there was only cleft palate in one of the cases, and both cleft palate and lip in the others. To date, there have been a large number of recognized syndromes, each of which is rare, that includes cleft lip and/or palate as a single feature. Of these, about 60% are manifestations of mutant genes and 40% do not appear to be familial. Some mutant genes can cause isolated cleft palate in some cases and cleft lip in others, with or without cleft palate. In other cases, isolated cleft palate is visible but not cleft lip. It has been estimated that less than 3% of all cases of cleft lip and/or palate represent a syndrome of some kind and that those with a genetic basis are more likely to have isolated cleft palate than cleft lip with or without cleft palate. There is strong evidence that secondary cleft palate is generally different from primary cleft palate and lip both developmentally and genetically during the embryonic period. Genetic findings in family studies showed that the incidence of isolated cleft palate increased in siblings of patients with isolated cleft palate, but the incidence of cleft lip did not increase. Genetic aberrations are known predisposing factors for syndromic and nonsyndromic CLPs. Structural and/or numerical CAs are associated with the OFCs. Changes in the structure or number of chromosomes disrupt the genetic balance. Such genetic changes lead to malformations of the genes responsible for lip and palate development, such as CL, CP, or both. Many studies have shown that orofacial clefts are associated with structural and numerical CAs or that they occur incidentally in Down, Patau, Edward and Klinfelter syndromes.
In the present study, numerical and structural damage was found in 4 of 10 patients, and two of these patients had 47, XXY, one had 47, XX, del(22q12>qter) or partial trisomy 22q12, and one had inv(9)(p11;q13). With this, the chromosome structure of the other 6 cases was normal. This suggests that in addition to gene dosage, other mechanisms such as genetic and/or environmental interactions and, possibly, imprinting may be important in determining the phenotypic outcome of patients with 22q12 duplication. With this, the sex ratio was 3/7 (male: female). It appears more in female cases than in males. This contradictory situation may be due to the small number of cases. Worldwide, it has been reported that CL/P is more common in men, while CP is more common in women. The sex ratio of CLP in the Caucasian population is 2:1 (male: female) [6]. A change in chromosome number can lead to abnormal genetic material that disrupts the embryonic development process. There are some autosomal and sex chromosome aberrations, albeit in small numbers, associated with oral abnormalities. In the present study, we reported 47, XXY karyotype (Klinefelter Syndrome=KS) in two boys. The seventeen-day case had phenotypic abnormalities such as CLP, droopy ears, hypertelorism, and unilateral cryptoorchidism. The other three-year-old case had unilateral cryptorchidism and mild mental retardation. The parents of both cases were under the age of thirty. Most newborns with KS have small, stiff testicles and varying symptoms of androgen deficiency, including gynecomastia, hypogonadism and infertility, short stature microcephaly, hypertelorism, flat nasal bridge, fifth finger clinodactyly, bifid uvula, heart defect, radioulnar synostosis, genu valgum, and similar clinical abnormalities [7,8]. However, oral anomalies are not among the phenotypic findings of XXY syndrome. Nevertheless, it has been suggested that the loss or addition of an X chromosome may affect the shape of the skull base and thus the measurement of facial prognathism [9,10]. Thus, in a very recent study, it was reported that XXY karyotype in one patient with OFC [11]. Similarly, one study reported a newborn baby with a 48, XXXY/46, XY karyotype with a cleft palate [12-16]. Mental retardation, cryptorchidism, radioguinal synostosis, clinodactyly, chest deformity and other bone anomalies, strabismus and cleft palate abnormalities have been reported in patients with XXXXY syndrome [17,18]. addition, congenital cleft palate anomaly was reported in two cases of 48, XXXY/46, XY mosaic in the past [12,19]. Although the relevance of such a difference is unknown due to the rarity of these cases, it is possible that an unusual phenotype of our case, cleft palate may be related to its karyotype.
The structural or numerical chromosomal changes may disrupt the functioning of the gene and cause malformations in the development of the lip and palate [20,21]. In the present study, we also found deletion and inversion type structural CA. The patient with only cleft palate had two normal complete chromosome 22 in addition to del(22)(q12 > qter) or an extra 22q12 region, and the other patient with both cleft palate and lip hadinv(9). Microdeletions or microduplications occur very frequently in the region of chromosome 22q11.2 located in the proximal region. Similarly, several autosomal injuries are known to cause oriofacial abnormalities. In addition to 22q11.2 deletions and duplications, other chromosomal abnormalities are also associated with CLP, often occurring in complex syndromes such as the chromosome 4p16.3 deletion in Wolf-Hirschhorn syndrome [22]. Similarly, genetic heterogeneity of chromosome regions 6p23, 2q13 and 19q13.2 and loci 4q25-4q31.3 and 17q21 has been reported in patients with cleft palate. CLP-associated microdeletions have also been reported in chromosome regions 20p12.3, 5q35.2-q35.3, 14q22.1-22.2, 4q21, 6p25.3 and 16p13.3 [23-29]. Also, it has been reported that bilateral cleft lip and bilateral thumb polydactyly develop as a result of deletion of some genes in chromosome regions 7p14.1, 4q32 and 4q34 [30,31]. Although there are still a few candidate genes and molecular pathways, we do not have a definitive mutation to explain the genetic background of most cases. Recent findings suggest that mutations or cytogenetic disruptions affecting specific cis-acting regulatory regions may play a decisive role. It has been reported that the inheritance pattern of CLP is compatible with the recessive single gene model [32].
Many genes contribute to the incidence of isolated syndromic cleft lip/palate cases. Although CLP formation is known to be associated with a number of genes such as transmembrane protein 1 and GAD1, it has also been found to be associated with mutations in the HYAL2 gene [33,34]. Especially sequence variants in IRF6, PVRL1 and MSX1 genes, and BMP4, SHH, SHOX2, FGF10 and MSX1 genes involved in midface morphogenesis have been widely reported [35].
Chromosome 22 is the second smallest chromosome in the human genome. The long arm of this acrocentric chromosome contains protein-coding genes. These disorders are common, with a prevalence of 1:2000, such as velocardiofacial syndrome (22q11 deletion syndrome, DiGeorge syndrome). Extra copies of the proximal region of chromosome 22q are known to cause cat eye syndrome, 22q11.2 duplication. Der(22) syndrome and cat eye syndrome are rare conditions characterized by increased copy number of the 22q11 region.
The clinical findings of patients with this deletion show a wide spectrum. The reason for the wide phenotypic variation is unknown. It has also been suggested that patients with 22q11.2 deletion are susceptible to other syndromes. The reason for the wide phenotypic variation is unknown. Syndromes associated with recurrence of the 22q11.2 region show phenotypic variability ranging from severe abnormality to mild features or even a completely normal phenotype [36,37].
The most frequently reported symptoms in this duplication syndrome are mental retardation/learning difficulties, delayed psychomotor development, growth retardation, and muscle hypotonia. Other most common dysmorphic features are hypertelorism, broad flat nose, micrognathia, velopharyngeal insufficiency, dysplastic ears, epicanthal folds, and downward sloping palpebral fissures. Less common symptoms are congenital heart malformation, visual and hearing impairment, seizures, microcephaly, ptosis, and urogenital abnormalities. 14 It has been reported that the prenatal phenotypes of the repeat sequences in the 22q11.2 region are different and this may be related to gene function [38].
In the literature, haploinsufficiency or triplosensitivity score would further support pathogenicity assessment of chromosomal repeat sequences to evaluate whether the phenotypic differences occurring in patients with 22q11.2 variants are evidence that these genes/regions are dose sensitive. Although most of the dozens of genes in the 22q11.2 region are well characterized, most of the expression mechanism of 22q11 is not yet known. Data on the penetration of this number of repetitive sequences are quite lacking. Some patients appear phenotypically normal, while others with the same genotype have mild to severe abnormalities. Therefore, more evidence needs to be gathered regarding the genotype-phenotype contributions of different 22q11.2 duplicated regions. It has been reported that proximal region-associated variants of abnormal 22q11.2 repeat sequences show more severe clinical phenotypes, while those associated with central and distal regions show milder or even normal features [39]. Considering all the phenotypic differences, we think that anomalies such as cleft palate and lip can be considered as an indicator for 22q aberrations.
Chromosome inversions are a relatively common structural alteration. We found 46, XY,i nv(9)(p11;q13) chromosomal aberration in one patient. Inv(9) is one of the most common (1-3%) balanced structural chromosomal abnormalities and is considered a normal familial variant. However, it has been reported in individuals with recurrent miscarriages, mild growth retardation, craniofacial malformations, undescended testicles, skeletal malformations, mental retardation, and hermaphroditism [40-42]. It seems difficult to decide whether this inversion is a chromosomal abnormality or a polymorphic variant of the chromosome. Geneticists have sought the answer to this question for years. A different dysmorphic symptom was described in many cases with del(9)(pter-p22 or 21). While malformations of facial features are more common in these patients, other congenital malformations are relatively rare and mild. Rarely, cleft palate, diaphragmatic hernia, hydronephrosis, radiological anomalies of the ribs and vertebrae are also seen [43]. Similarly, pericentric inversion of chromosome 9 was reported in two patients with oriofascial cleft in a very recent study [11]. More research is needed to definitively state the relationship of inv(9) to cleft palate.
Conclusion
22q12 is associated with other serious congenital anomalies such as cleft palate. But, it is still unknown whether trisomy cytogenetic studies contribute to finding genes involved in the unknown genetic etiology of CLP. Although oral anomalies are not among the phenotypic findings of XXY syndrome, our findings suggest that an excess of one X chromosome may affect the development of cleft palate and lip. At the same time, our findings support that the dosage of some genes in the 22q12 region contributes to CLP, indicating that it may be effective in better characterizing the complex genotype-phenotype relationship of the disease. Understanding the etiology of OFCs is also important for developmental biology. Therefore, the 22q12 region may help us better understand the complex pathogenesis of CLP. However, molecular genetic analyzes in patients with CLP will help to fully reveal the genetic etiology of the disease. Additionally, it is necessary to provide medical-genetic counseling to parents of children with CLP.
Author Statements
Consent
Informed consent was obtained from the patient's parents for publication of this case report and accompanying images.
References
- Rahimov F, King OD, Leung DG, Bibat GM, Emerson CP, Kunkel LM, et al. Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkers. Proc Natl Acad Sci USA. 2012; 109: 16234-9.
- Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009; 374: 1773-85.
- Wyszynski DF, Beaty TH. Review of the role of potential teratogens in the origin of human nonsyndromic oral clefts. Teratology. 1996; 53: 309-17.
- Yigit AK, Oguz SS, Dilmen U. Collecting and observing the growths of the cases with cleft lip and cleft palate. The J Gynecol-Obstet Neonatol. 2015; 12: 80-2.
- Junichi AZ, Kohama Gi SM. Cytogenetic studies in patients with cleft lip and/or cleft palate (IV), lap. Hum Genet. 1978; 23: 161-6.
- Mossey PA, Chapter LJ 12. Epidemiology of oral clefts: an international perspective. In: Wyszynski DF, editor. Cleft lip and palate. From origin to treatment. Oxford University Press. 2002; 27-58.
- Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet. 2004; 364: 273-83.
- Visootsak J, Aylstock M, Graham JM Jr. Klinefelter syndrome and its variants: an update and review for the primary pediatrician. Clin Pediatr. 2001; 40: 639-51.
- Ingerslev CH, Solow B. Sex differences in craniofacial morphology. Acta Odontol Scand. 1975; 33: 85-94.
- Froland A. Klinefelter’s syndrome. Clinical, endocriological and cytogenetical studies. Dan Med Bull Suppl. 1969; 16: 1-108.
- Sabnis AS, Natrajan S. Cytogenetic evaluation of orofacial clefts. Natl J Clin Anat. 2021; 10: 199-204.
- Hur M, Cho HC, Lee KM, Park H, Lee SY, Kim KN, et al. Cleft palate in a rare case of Variant Klinefelter syndrome with 48,XXXY/46,XY mosaicism. Cleft Palate Craniofac J. 2009; 46: 555-7.
- Gupte GL, Kotvaliwale SV, Mahajan JV, Kher AS, Kanade SP, Bharucha BA. 48 XXXY variant of Klinefelter syndrome. Indian Pediatr. 1995; 32: 798-801.
- Kruse R, Guttenbach M, Schartmann B, Schubert R, van der Ven H, Schmid M, et al. Genetic counseling in a patient with XXY/XXXY/XY mosaic Klinefelter’s syndrome: estimate of sex chromosome aberrations in sperm before intracytoplasmic sperm injection. Fertil Steril. 1998; 69: 482-5.
- Velidedeoglu HV, Demir Z, Bozdogan MN, Coskunfirat OK, Kurtay A, Türkgüven Y. Uncommon Klinefelter’s variant (49,XXXXY) with cleft palate. Ann Plast Surg. 1997; 39: 213-5.
- Visootsak J, Aylstock M, Graham JM Jr. Klinefelter syndrome and its variants: an update and review for the primary pediatrician. Clin Pediatr. 2001; 40: 639-51.
- Fraser JH, Boyd E, Lennox B, Dennison WM. A case of XXXNY KlinefcIter’s syndrome. Lancet. 1961; 2: 1064-7.
- Mcenery ET, Brenneman J. MultipIe faciaI anomahes. J Pediatr. 1937; II: 468.
- Abdelmoula NB, Amouri A, Portnoi MF, Saad A, Boudawara T, Mhiri MN, et al. Cytogenetic and fluorescence in situ hybridization assessment of sex-chromosome mosaicism in Klinefelter’s syndrome. Ann Genet. 2004; 47: 163-75.
- Hirakawa M, Adachi K. Cytogenetic studies in 100 patients with cleft lip and cleft palate. J Kyushu Dent Soc. 1970; 24: 73-85.
- Azumi J, Kohama G, Sasaki M. Cytogenetic Studies in Patients with Cleft Lip and/or Cleft Palate (Screening Studies of Chromosomes. Hum Genet. 1978; 23: 161-6.
- Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet. 2008; 148: 246-51.
- Williams ES, Uhas KA, Bunke BP, Garber KB, Martin CL. Cleft palate in amultigenerational family with amicrodeletion of 20p12.3 involvingBMP2. Am J Med Genet A. 2012; 158A: 2616-20.
- Sahoo T, Theisen A, Sanchez-Lara PA, Marble M, Schweitzer DN, Torchia BS, et al. Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am J Med Genet A. 2011; 155A: 1646-53.
- Peredo J, Quintero-rivera F, Bradley JP, Tu M, Dipple KM. Cleft lip and palate in a patient with 5q35.2-q35.3 microdeletion: the importance of chromosomal microarray testing in the craniofacial clinic. Cleft Palate Craniofac J. 2013; 50: 618-22.
- Bhoj E, Halbach S, McDonald-McGinn D, Tan C, Lande R, Waggoner D, et al. Expanding the spectrum of microdeletion 4q21 syndrome: a partial phenotype with incomplete deletion of the minimal critical region and a new association with cleft palate and Pierre Robin sequence. Am J Med Genet A. 2013; 161A: 2327-33.
- Chen CP, Lin SP, Chern SR, Wu PS, Su JW, Wang W. A boy with cleft palate, hearing impairment, microcephaly, micrognathia and psychomotor retardation and a microdeletion in 6p25.3 involving the DUSP22 gene. Genet Couns. 2013; 24: 243-6.
- Demeer B, Andrieux J, Receveur A, Morin G, Petit F, Julia S, et al. Duplication 16p13.3 and the crebbp gene: confirmation of the phenotype. Eur J Med Genet. 2013; 56: 26-31.
- Lumaka A, Van Hole C, Casteels I, Ortibus E, De Wolf V, Vermeesch JR, et al. Vari ability in expression of a familial 2.79 Mb microdeletion in chromosome 14q22.1-22.2. Am J Med Genet A. 2012; 158A: 1381-7.
- Calcia A, Gai G, Di Gregorio E, Talarico F, Naretto VG, Migone N, et al. Bilaterally cleft lip and bilateral thumb polydactyly with triphalangeal component in a patient with two De novo deletions of HSA 4q32 and 4q34 involving PDGFC , GRIA 2 , and FBXO 8 genes. Am J Med Genet A. 2013; 161: 2656-62.
- Younkin SG, Scharpf RB, Schwender H, Parker MM, Scott AF, Marazita ML, et al. A genome-wide study of de novo deletions identifies a candidate locus for nonsyndromic isolated cleft lip/palate risk. BMC Genet. 2014; 15: 24.
- Carinci F, Scapoli L, Palmieri A, Zollino I, Pezzetti F. Human genetic factors in nonsyndromic cleft lip and palate: an update. Int J Pediatr Orl. 2007; 71: 1509-19.
- Sandoiu A. Scientists find genetic mutation that causes cleft lip and palate, heart defects. Med News Today. 2017; 31.
- Kanno K, Suzuki Y, Yamada A, Aoki Y, Kure S, Matsubara Y. Association between nonsyndromic cleft lip with or without cleft palate and the glutamic acid decarboxylase 67 gene in the Japanese population. Am J Med Genet A. 2004; 127A: 11-6.
- Cox TC. Taking it to the max: the genetic and developmental mechanisms coordinating midfacial morphogenesis and dysmorphology. Clin Genet. 2004; 65: 163-76.
- Rump P, de Leeuw N, van Essen AJ, Verschuuren-Bemelmans CC, Veenstra-Knol HE, Swinkels ME, et al. Central 22q11.2 deletions. Am J Med Genet A. 2014; 164A: 2707-23.
- Pinchefsky E, Laneuville L, Distal SM. 22q11.2 microduplication: case report and review of the literatüres. Child Neurol Open. 2017; 4: 2329048X17737651.
- Cao P, Zhu X, Gu L, Liu W, Li J. Analysis of related phenotype of prenatal cases with copy number variations in various region of 22q11.2. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2021; 38: 1055-9.
- Xue J, Shen R, Xie M, Liu Y, Zhang Y, Gong L, et al. 22q11.2 recurrent copy number variation-related syndrome: a retrospective analysis of our own microarray cohort and a systematic clinical overview of Clin Gen curation. Transl Pediatr. 2021; 10: 3273-81.
- Demirhan O, Pazarbasi A, Suleymanova-Karahan D, Tanriverdi N, Kilinc Y. Correlation of clinical phenotype with a pericentric inversion of chromosome 9 and genetic counseling. Saudi Med J. 2008; 29: 946-51.
- Lourenço GJ, Silva PMR, Bognone RAV, De Souza RA, Delamain MT, Lima CS. Inherited pericentric inversion of chromosome 9 in acquired hematological disorders. Ann Hematol. 2007; 86: 465-7.
- Ramegowda S, Savitha MR, Krishnamurthy B, Doddaiah N, Prasanth SN, Ramachandra NB. Association between pericentric inversion in chromosome 9 and congenital heart defects. Int J Hum Genet. 2007; 7: 241-8.
- Schinzel A. Catalogue of unbalanced chromosome aberrations in man. De Gruyter. 1983.