
Abstract
Cardiac dysfunction in cirrhosis of liver remains dormant due to hyperdynamic circulatory state even with the severe stage of cirrhosis. This in actuality is worsening of the cardiac functions. The decline in diastolic functions, inotropic and chronotropic functions and cardiac hypertrophy all occur simultaneously in the setting of an absent organic cardiac disease. The Cirrhotic cardiomyopathy has pertinent findings in its loop comprising of impaired contractile reaction to stress stimuli and electrophysiological abnormalities along with prolonged QT interval. The disruption in β-adrenergic receptor signalling, altered composition of cardiomyocyte membrane lipids plus biophysical properties, ion channel defects and enhanced cardiodepressant factors attributed to hormones are the pathogenic assailants. The hindrance to diagnose cirrhotic cardiomyopathy mainly lies in unavailability of a stark specific diagnostic test nevertheless; an echocardiogram is favourably used to follow deteriorating diastolic functions and the E/e' ratio thus giving insight to the progression of disease. The severity of cirrhosis is linked in parallel with ensuing cirrhotic cardiomyopathy which substantially impairs arterial blood volume. So, in case of any hemodynamic stress, a heart bearing cirrhotic cardiomyopathy retorts with diminished cardiac response which may cause renal hypoperfusion leading to renal failure. The management is mainly symptomatic where only the liver transplantation could play an imperative role in correction of the cardiac functions.
Keywords: Cirrhotic cardiomyopathy; Cardiac dysfunction; Left ventricular diastolic dysfunction; Arterial blood volume; Hepatorenal syndrome
Abbreviations
CCM: Cirrhotic Cardiomyopathy; QTc: Corrected QT interval; SNS: Sympathetic Nervous System; RAAS: Renin Angiotensin Aldosterone System; CAIDS: Cirrhosis-Associated Immune Dysfunction Syndrome; NO: Nitric Oxide; CO: Carbon monoxide; LV: Left Ventricle; SVR: Systemic Vascular Resistance; BDL: Bile Duct Ligation; PWCP: Pulmonary Wedge Capillary Pressure; PRAL: Plasma Renin Activity; TGF β: Transforming Growth Factor ?; IVRT: Increased Isovolumic Relaxation Time; DT: Deceleration Times; TDI: Tissue Doppler Imaging; CAMP: Cyclic Adenosine Monophosphate; PKA: Protein Kinase; ECS: Endocanabinoid System; iNOS: inducible Nitric Oxide Synthase; L-NMMA: N Omegamonomethyl-larginine; NGL: Nitro-arginine Methyl Ester; HO: Haem Oxygenase; CGMP: Cyclic Guanosine Monophosphate; MAPKs: Mitogen-Activated Protein Kinase; HRS: Hepatorenal Syndrome; ANP: Atrial Natriuretic Peptide; BNP: B Type Natriuretic Peptide; GLS: Global Longitudinal Strains
Introduction
Cirrhosis is a chronic state of liver caused by various aetiologies characterized by altered parenchyma and distorted hepatic vascular architecture consequent to chronic tissue fibrosis and regenerative nodules [1,2]. Globally, cirrhosis has become an emergent cause of mortality. Apart from the known complications of cirrhosis like ascites, hepatic encephalopathy, upper GI bleeding and Coagulopathy, cardiac involvement in the form of Cirrhotic Cardiomyopathy (CCM) has recently gained attention as the commonest cause of post liver transplant mortality [3].
Cardiac dysfunction was established in cirrhosis of liver half a century ago when cardiovascular changes like hyperdynamic circulation, high cardiac output, reduced peripheral vascular resistance and reduced blood pressure were described [4,5]. Consequent pathological evidence of cardiac dysfunction in cirrhosis of liver was cardiac hypertrophy, edema of cardiomyocyte without valvular heart disease, coronary artery disease and hypertension [6]. Initially, it was believed that the cardiac dysfunction is directly consequent to alcohol toxicity and was termed as latent alcoholic cardiomyopathy [7]. The decreased hemodynamic retort to either of the physiologic (exercise) or pharmacologic strain even with an elevated basal cardiac output which was witnessed in a few trials on human and animal models of non-alcoholic cirrhosis [8,9]. This review spotlights on definition, pathophysiology, clinical significance, diagnosis and management of CCM.
Definition of CCM
CCM is defined as impaired cardiac functions without any organic cardiac disorder pertaining to diminished cardiac contractility when stimulated (physiological / pharmacological) and adapted relaxation during diastole with electrophysiological abnormalities in patients with cirrhosis [10-13]. The consensus diagnostic criteria are shown for CCM in (Table 1).
![]()
Cirrhotic Cardiomyopathy
Cardiac dysfunction in patients suffering from cirrhosis character–ized by impaired contractile responsiveness to stress and/or altered diastolic relaxation with associated electrophysiological abnormali�ties without underlying known cardiac disease.
Diagnostic Criteria
Systolic dysfunction
Blunted increase in cardiac output with exercise, volume challenge or pharmacological stimuli
Resting E > 55
Diastolic dysfunction
E/A <� 1
Prolonged deceleration time (200 msec)
Prolonged isovolumetric relaxation time (80 msec)
Supportive criteria
Electrophysiological abnormalities
Chronotropic incompetence
Electromechanical uncoupling
Prolonged QTc interval
Enlarged left atrium
Increased myocardial mass
Increased BNP, pro-BNP
Increased Troponin I
Table 1: Proposal for diagnostic and supportive criteria for cirrhotic cardiomyopathy as proposed by World Congress of Gastroenterology (2005) Montreal.
Epidemiology
The data on actual prevalence of CCM is scanty because of its silent nature with near normal cardiac functions unless it becomes unmasked due to stress in form of upper GI bleeding and sepsis. The estimated prevalence of CCM is between 40 to 50% in advance stage irrespective to aetiology of cirrhosis [14]. In previous studies on patients waiting for liver transplantation, 50% patients with decompensated cirrhosis had impaired cardiac functions. 7% -21% of post-transplant deaths are attributable to cardiac failure [15,16]. Majority of the patients with advanced cirrhosis have at least one component of CCM either diastolic dysfunction or prolonged QTc [17].
Possible Commencers of CCM
Patients of cirrhosis with established portal hypertension bear several structural modifications in the heart (cardiac wall thickness and chamber dilatation). Some anatomical defects observably are adaptations to hyperdynamic circulation. The initiation of CCM is linked to the following factors;
Portal hypertension
Portal hypertension with hepatic decompensation is associated with severity of CCM. RuÍz-del-Árbo et al. have demonstrated the associated extent of CCM and advance cirrhosis [18]. The study further revealed dysfunction of diastole in cirrhotics having severe hepatic decompensation and ascites. Chronotropic and left ventricular systolic response to peripheral vasodilatation and Sympathetic Nervous System (SNS) is impaired in cirrhotics [18]. A recent study comparing cirrhotic with or without cardiomyopathy has shown a positive correlation between advanced cirrhosis and all components of cardiomyopathy [19].
Changes in circulation
Before the advanced cirrhosis, hyperdynamic circulation compensates splanchnic vasodilatation but as the cirrhosis advances, steadily increasing vasodilatation reduces the arterial blood volume. Decreased blood volume triggers the Renin Angiotensin Aldosterone System (RAAS) and SNS [20]. These circulatory changes cause both structural (chamber dilatation) and functional cardiac defects in cirrhosis [21].
Inflammation and cytokines
Impaired immunity (defective humoral and complement mediated immunity) is multifactorial which increases risk of infections among cirrhotics [22]. Inflammatory cytokines like reactive nitrogen species and oxygen species released in decompensated cirrhosis have been linked to cardiac dysfunction. The postulated inciters of immune dysfunction are bacterial overgrowth, increased porosity of gut and translocation of bacteria into circulation from gut. Bacterial lipopolysaccharides and their DNA permeate gut into circulation and activate monocytes and lymphocytes to release various cytokines. Various cytokines exert inhibitory effects on left ventricular systole and extracellular matrix (cardiac remodeling) [23-26].
Pathophysiology
Vascular impairment
Changes in systemic vascular resistance, cardiac output and splanchnic circulations have been studied both in experimental and human cirrhosis. These changes are related to advance liver disease. Portal hypertension at the level of pre-sinusoids in rats showed splanchnic arterial vasodilation, decreased myocardial contractility to isoproterenol or nitroprusside with hampered calcium signaling mechanism in myocyte. The above observation concluded that portal vascular alteration and Porto systemic shunting eventuates as cirrhotic cardiomyopathy which is partially independent of liver disease itself [27-29]. Contrarily, portal hypertension in sinusoidal architecture is a combination of increased sinusoidal resistance (fibrotic disturbance of parenchyma) and a dynamic (defective contractile nature of hepatic stellate cells and myofibroblasts) [30]. Hepatic stellate cells and myofibroblasts contractility depends on various vasoactive mediators like Nitric Oxide (NO), endothelins and prostaglandins. The cirrhotic state impairs production of NO due to increased caveolin expression [31,32]. Peripheral and splanchnic vasodilation occurs when NO increases. Endogenous cannabinoids and Carbon Monoxide (CMO) leads to splanchnic vasodilation [33]. Accumulation of endogenous cannabinoid, NO and CMO could have negative inotropic effect and contributes in diastolic dysfunction [34,35].
Redistribution of blood volume
Blood volume increases in cirrhosis, even before ascites develop. There is an evident redistribution of expanded plasma volume where blood mostly diverts to splanchnic bed causing relative decrease in central circulation [36]. This volume redistribution among cirrhotics is related to the degree of decompensation of liver.
Vascular remodeling
Arterial compliance is always dependent on arterial vasodilation and nature of the arterial wall. As the cirrhosis progress, vascular wall thickness decreases subsequently decreasing total vascular wall area [37]. There is reduced vascular tone in cirrhotic patients due to decrease smooth muscle mass explained by increased production of NO, endothelial dysfunction and increase turnover of extracellular matrix. Vascular remodeling and modified arterial compliance in cirrhosis has been linked to over expression of large conductance and a subunit of calcium activated K channel [33].
Pathogenesis of CCM
CCM is characterized by sequential cardiac alterations responsible for its signature clinical implications. Following pathogenic mechanisms have been suggested to various components CCM. The salient components are depicted in (Figure 1).
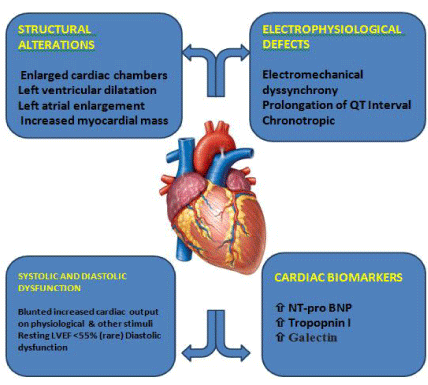
Figure 1: Salient Components of Cirrhotic Cardiomyopathy.
Systolic dysfunction
The data on cirrhotics (both animals and humans) evidences that systolic functions remain normal or even above normal at rest. The underlying systolic dysfunction becomes unmasked whenever there is physiologic (exercise) or pharmacologic (dobutamine infusion) stimulus [18]. Blunted cardiac responsiveness to volume, postural changes, exercise or pharmacological stimulus has been recognized in cirrhotics.
Physiologic stress: The postural changes and exercise produce cardiac dysfunction among cirrhotics. When cirrhotic patients were kept standing for 5 minutes, in spite of increased HR, LV end systolic volume, cardiac index and peripheral systemic resistance decrease [37,38]. Exercise in cirrhotics when compared with normal subjects has shown abnormal LV response (diminished increase in CO and ejection fraction) [11]. Patient with cirrhosis during exercise have shown unaffected cardiac index and ventricular filling pressure [7]. Non invasive assessment of ventricular contractile performance in cirrhotics at rest and during exercise can be done by measuring systolic time interval. It is assessed from concurrent tracings of carotid artery pulse, phonocardiogram and electrocardiogram). Increased systolic time interval prolongs total electromechanical systole in cirrhotics. Impaired adrenergic drive causes electromechanical delay thus increasing systolic time [13].
Effect of ascites: The effect of ascites on cardiac dysfunction has been evaluated in various studies. Pozzi et al. [9] showed that ascites and therapeutic paracentesis do not affect cardiac dysfunction. Wong et al. [39] have shown end systolic volume increase due to sodium retention causing compromised cardiac contractility even before ascites ensues. The effect becomes more important when ascites develop despite reduction of both pre and after load [10].
Pharmacologic stress: Blunted cardiac response to various pharmacologic agents has been reported in cirrhotic patients. Epstein M et al. [40] showed failure in increasing cardiac output in cirrhotics when angiotensin II was infused despite increased Pulmonary Wedge Capillary Pressure (PWCP) and normalization of SVR. Terlipressin infusion also has the same effects in patients with cirrhosis [41]. Above studies concluded that if after load normalizes, it may help in detection of LV dysfunction. Moreover dobutamine (β1-adrenergic receptor agonist) in cirrhotics showed only a small rise in stroke volume (negative inotropism). Comparatively, more amount of isoproterenol is required to enhance heart rate in cirrhotics versus healthy controls (negative chronotropism) [42,43].
Diastolic dysfunction
The diastolic dysfunction even heralds systolic dysfunction in cirrhosis. There is defective ventricular relaxation (premature passive (E) and late (A) components) during ventricular filling phase, which subsequently increases atrial pressure and isovolumetric relaxation time [44,45].
Impaired myocardial relaxation: Defective calcium exchange through the sarcoplasmic reticulum is linked with impaired myocardial relaxation explains diastolic dysfunction. The elasticity of relaxed striated muscle is dependent on titin which determines diastolic stiffness. Myocardial extracellular matrix is also affected with alteration in diastolic function. Experimental models of Bile Duct Ligated Animals (BDL), show decreased phosphorylation of titin increasing passive tension [46]. Histopathological evidence of diastolic dysfunction has shown interstitial and cardiomyocyte fibrosis along with impair pigmentation and myocyte vacuolization [47]. Chances of supraventricular arrhythmias like atrial fibrillation increase by augmented atrial volume consequent to impaired ventricular filling.
Myocardial hypertrophy and myocardial fibrosis: High salt intake among animals has shown concentric LV hypertrophy related to excessive production of aldosterone [48]. LV hypertrophy in cirrhotics is mainly responsible for diastolic dysfunction [49]. Among structural changes thickness of heart walls has been associated with cirrhosis of liver [50]. Pathogenesis of myocardial fibrosis and aldosterone is linked for stimulating pro inflammatory cytokines like transforming growth factor β (TGF β) [51]. Patients having ascites (decompensated cirrhosis) with raised plasma epinephrine and Plasma Renin Activity (PRA) showed more LV hypertrophy [34,52] than patients without ascites having normal PRA [18].
Echocardiographic parameters: The diastolic dysfunctions can be determined both by invasive (measuring pulmonary capillary wedge pressure plus end diastolic pressure) and non invasive (echocardiography) methods. Reduced SVR and central hypovolemic state in CCM explains increased cardiopulmonary pressure with normal mean left atrial or PCWP [53]. Salient echocardiographic assessments in CCM includes left atrial dilatation with raised left atrial volume, increased LV diameter without an increase in LV volume and thickness of posterior wall of LV and inter cardiac septum. Increased Isovolumic Relaxation Time (IVRT), decreased peak E velocity (early rapid filling phase), prolong Deceleration Times (DT) of the E wave, and finally peak A velocity increased (atrial contraction during late diastole) have been shown on echocardiography [54].
Ascites also affects diastolic dysfunction where reduced E/A ratio (echocardiographic evidence of dysfunction of diastole) have been found. Alternatively, other parameters of diastolic dysfunction like prolong IVRT and DT remain independent to ascites development [39]. Diastolic dysfunction (normal, pseudonormal and restrictive patterns) is assessed by Doppler echocardiogram where altered E/A ratio indicate diastolic dysfunction. E/A ratio determined by Doppler echocardiography have got limitations for inability to exactly differentiate between pseudo normal versus normal filling pattern. Tissue Doppler Imaging (TDI) is a latest technique where tissue velocity of mitral annulus (e') at the basal and septal angle is assessed which is considered more sensitive indicator of diastolic dysfunction. Diastolic dysfunctions are considered when lateral e' <10 cm/s, septal e' < 8 cm/s, mitral inflow patterns and LA volume index = 34 mL/m2 (Figure 2) [55].
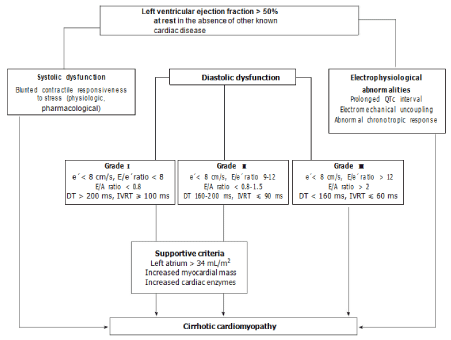
Figure 2: Algorithm for the diagnosis of cirrhotic cardiomyopathy. Figures shows systolic dysfunction (Blunted cardiac response to stress stimuli) Diastolic
dysfunction as assessed by TDI, Electrophysiological abnormalities and supportive criteria.
CCM: Cirrhotic Cardiomyopathy; EF: Ejection Fraction; LVDD: Left Ventricular Diastolic Dysfunction; TDI: Tissue Doppler Imaging; e': Peak early diastolic velocity
at the basal part of the septal and lateral corner of the mitral annulus; E/e' ratio: Peak E-wave transmitral/early diastolic mitral annular velocity; E/A ratio: Early
diastolic mitral inflow velocity/late diastolic velocity; DT: Deceleration Time; IVRT: Isovolumic Relaxation Time [55], [copyright© 2015 Baishideng Publishing Group
Inc. Reused with the kind permission of World Journal of Gastroenterology].
Severity and correlation: Severity of diastolic dysfunction is usually classified according to average E/e' ratio from mild grade 1 to severe grade 3. Most cirrhotics have mild grade 1 or moderate grade II diastolic dysfunction. Diastolic dysfunctions have been well correlated with severity of liver disease [19] and high MELD score [18]. RuÍz-del-Árbo et al. [18] have shown an association of diastolic dysfunction severity and derangement of systolic indices at rest in patients with CCM. Karagiannakis et al. [56] showed correlation between E/e' ratio (diastolic dysfunction) and serum bacterial endotoxin marker (lipopolysaccharide-binding protein).
Electrophysiological aberration
Cirrhosis of liver brackets together various electrophysiological irregularities including electromechanical uncoupling, prolongation of QT interval, and chronotropic ineffectiveness [57].
Prolongation of QT interval: Prolong QT interval (>0.44 S) has been observed with portal hypertension with and without cirrhosis. Its frequency among cirrhotics is about 30-60% correlated to the worsening of liver disease [58]. Heart rate affects QT interval so rate related correction is always required to assess exact interval called corrected (QTc). For calculation of QTc in cirrhotics, Fridericia formula [59] is applied. Ventricular repolarization responsible for QT interval varies in its duration to even the slightest change in portal pressure among cirrhotics. Loss of K channel on plasma membrane and their dysfunctions are responsible for delayed ventricular repolarization eventually leading to prolong QT interval [60-62]. Lengthen QT interval is also elucidated by the hyperactivity of sympathoadrenergic discharge as evidenced by high circulating levels of noradrenalin. Autonomic dysfunction [63] along with exposure of different cytokines through portosystemic shunts to heart are also proposed mechanism for prolong QT interval [64,65]. It is worth noting that patients undergoing liver transplantation are found to have prolonged QTc interval (= 500 ms). QTc gets corrected after transplantation in about 50% of the patients [66]. Chronic β-blocker administration has shown reduction in prolong QT interval [67]. Upper GI bleeding and TIPS are associated with prolong QT interval [68]. Patients with prolong QT interval may have ventricular arrhythmias [55].
Electromechanical uncoupling: The dyssynchronus contractile response of systole to electrical signals is termed as electromechanical uncoupling. Electromechanical dyssynchrony has two components, preejection phase (time interval between ventricular depolarization and ventricular ejection) and LV ejection phase. Extended preejection time delay and electromechnical delay have been reported among cirrhotics both at rest and after isometric exercise, thus suggested electromechanical dyssynchrony [10]. Decrease concentration of L-type calcium channels are associated with defective excitation contraction coupling in animal models [66]. Cirrhotics with prolonged QT interval have more chances of electromechanical uncoupling. Dysfunctional K channels are associated with impaired excitation contraction coupling [69]. Electromechanical uncoupling is observed commonly in patients administered with terlipressin for variceal bleeding [45].
Chronotropic and inotropic incompetence: Incapability of heart to proportionally increase its rate in response to metabolic challenges is known chronotropic incompetence. Chronotropic response to dobutamine remains normal in early cirrhosis [70]. Chronotropic incompetence is observed in advance cirrhosis irrespective to underlying aetiology. Cirrhotics have revealed chronotropic incompetence in response to exercise, pharmacologic stimuli, paracentesis and infections [71]. Reduced sensitivity of activation in Sympathetic Nervous System (SNS) due to down regulation and desensitization to beta adrenergic receptors in sinoatrial node is the proposed mechanism for chronotropic incompetence [72].
Autonomic dysfunction
Autonomic dysfunction related to cardiovascular system frequently observed in decompensated cirrhosis with a reported incidence of autonomic neuropathy of 35% to 80%. Autonomic dysfunction is related to the stage of cirrhosis [73,74]. Heart rate variability, impaired sensitivity of baroreflex is well described autonomic dysfunctions [75,76]. Augmented Sympathetic tone in cirrhotic patients leads to excessive interaction of catecholamine (noradrenalin) which causes myocardial damage and down regulation of β-adrenergic receptor [77].
β-Adrenergic receptors dysfunction: Myocardial contractility is strictly modulated by the β adrenoceptors system. β adrenoceptors system is activated by catecholamine’s (adrenaline and noradrenalin) which are coupled with GS protein and causes an ultimate discharge of cAMP. The Gs protein system is also responsible for direct opening of ca channel on scarcolemma. cAMP is a well known second messenger which activates its dependent Protein Kinase (PKA). Activated protein kinase phosphorylates L-type Ca channel protein, tropopnin and ryanodine receptor which ultimately increases influx of calcium within myofibrils and causes contraction [55]. Experimental cirrhosis (cirrhotic rats) have shown impaired β adrenergic signal on cardiomyocyte. Several molecular derangements such as decrease density, desensitization, defective production of cAMP and adenylyl cyclase of β adrenergic receptors have been discovered in cirrhotic rats [78]. Down regulation of β adrenergic receptors were found on lymphocytes of decompensated cirrhotic patient which was correlated with cardiac contractility [79].
Altered cardiomyocyte membrane: Cardiomyocyte membrane fluidity changes due to alteration in the composition of plasma membrane both in animal models and human cirrhotic patients. Increase in plasma membrane cholesterol of cirrhotic patients lead to decrease fluidity and made erythrocytes rigid and impermeable [80]. These chemical changes in cardiomyocyte membrane impede with activation of β adrenoceptors system and calcium channels [81]. In the cirrhotic rat model reduced plasma membrane fluidity with abnormally expressed gene at the post receptor level of the adrenergic system [82] causes impaired second messenger (cAMP) signalling on infusion of isoproterenol [83]. Modified membrane fluidity hampers stimulation of cardiac muscarinic receptors responsible of pacemaker activity. Experimental cirrhosis has also shown defective responsiveness to muscarinic receptors [84]. Altered membrane cholesterol also causes modified activity of scarcolemma enzymes like Na+/Ca++ exchanger; ATPase and calcium pump ATPase leading to electrocardiograph changes [85].
Cardiac inhibitory factors
Following humoral factors with cardiosuppressant effect in experimental cirrhosis are;
Endocannabinoid (ECS): These are a group of molecules used for lipid signaling [86]. The ECS system (arachidonoyl ethanolamine and 2-arachidonoylglycerol) are found to be elevated in cirrhosis causing decrease intracellular cytosolic calcium concentration by inhibiting L type Ca++ channels on cardiomyocyte [87-89]. Reduced cardiac contractile response to isoprotenerol has been evidenced in the fibrocholestatic heart model.
Nitric oxide (NO): Excessive production of NO secondarily to enzyme inducible Nitric Oxide synthase (iNO) with a negative inotropic effect is reported in experimental studies [90]. BDL rat model has shown decrease cardiac contractility due to high level of iNO which is confirmed upon its reversal with administration of NOS inhibitor L-NMMA (N omegamonomethyl-l arginine) and L-NAME (NGL- nitro-arginine methyl ester) [91-93] Experimental evidence further suggested that increase activity of NO is related to different cytokines among cirrhotic animals. Effect of nitration of NO on cardiomyocyte has also been linked to CCM [93].
Carbon monoxide (CMO): CMO is a degradation product of haem which is catalyzed by enzyme Haem Oxygenase (HO). Endotoxemia, release of cytokines among cirrhotics stimulate HO which causes more production of CMO. Experimental cirrhotic model has shown unregulated activity of HO in the ventricles of BDL cirrhotic rats when compared to sham control rats [94]. Elevated level of CMO reduces cardiac contractility as evidenced by improvement in cardiac contraction after administration of zinc protoporphyrin which is an antagonist of CMO [93]. BDL cirrhotic rats have shown increase level of cyclic guanosine monophosphate cGMP in response to activation of the NO and CMO with their synthetic enzymes [92]. CGMP either by phosphorylation of G kinase protein or by reducing production of raynodine receptor ultimately causes decrease intra cellular calcium with cardiomyocytes [94].
Programme Cell Death and Cardiac Contractility
Programme cell death contributes in cardiac remodeling after heart failure. An impaired Na+/Ca++ exchanger on membrane of cardiomyocyte among cirrhotics leads to an excessive influx of calcium which initiates apoptosis in cardiomyocytes [95]. Recent evidence has shown the combined role of cardiosuppressants (CMO, NO, ECS) and apoptosis in CCM [96]. An extrinsic apoptotic pathway has been identified for impaired cardiac contractility in BDL cirrhotic mice [97]. Mitogen-Activated Protein Kinase (MAPKs) with its isoform p38-MAPK has been identified for apoptosis after ischemia in the experimental cirrhosis [98].
Clinical Features
CCM clinically remains asymptomatic initially until the physiological or pharmacologic stress has occurred which requires a high cardiac activity to preserve hemodynamic balance. Cardiac incompetence to combat with situations requiring increased cardiac output further increases with advancement of cirrhosis. In the advance stage of cirrhosis cardiac dysfunction and chronotropism become so enunciated that it even fails to increase cardiac output even without any stress conditions. Exercise tolerance decreases with left ventricular diastolic dysfunction [99]. Cardiac failure remains silent due to marked peripheral vasodilatation in cirrhosis. Overt heart failure and extensive pulmonary edema is reported in cirrhotic patients after TIPS and orthotropic liver transplantation [100,101].
Hepatorenal Syndrome (HRS)
CCM has been linked to the pathogenesis of HRS as it causes changes in effective circulatory volume, and considered as a sensitive indicator for development of HRS [18]. There is an ample data from longitudinal studies which has shown a correlation between systolic function and renal failure. Patients with tense ascites and normal baseline renal function who have developed HRS in their course of disease when compared to patients without HRS have shown more systolic dysfunction (lower baseline Cardiac output, stroke volume, left ventricular stroke work) and raised PRA and nor epinephrine level [102]. PRA and cardiac output are considered as independent predictors of HRS. Patient with severe CCM with normal renal functions are more prone to develop HRS [18]. Recent study has shown an important relationship between renal impairment and extent of systolic dysfunction [103].
Sepsis and SBP
Heart failure becomes evident in the case of sepsis due to decrease cardiac output and hypotension. The high level of inflammatory cytokines generates metabolic stress, which is responsible for circulatory failure [100,104].
Sudden death
Patients with cirrhosis of liver may have sudden cardiac death which has been reported as a clinical implication of CCM [100]. It is associated with QT interval prolongation which has been associated with ventricular arrhythmias. Upper GI bleeding also increases prolongation of QT interval [100].
Diagnosis of CCM
Diagnostic criteria for CCM were proposed by an expert panel in 2005 at the World Congress of Gastroenterology in Montreal (Table 1) which includes systolic, diastolic and electrophysiological components as given in (Table 1) [47,44]. Ruiz-del-Árbol L et al. [55] has recently proposed a useful diagnostic algorithm for CCM.
Laboratory Findings
Cardiac biomarkers
The elevation of Atrial Natriuretic Peptide (ANP) and B-type Natriuretic Peptide (BNP) as biomarkers have been found as an indicator of compromised myocardial contractility and impaired diastole among cirrhotics. ANP and BNP were included as supportive criteria of CCM.
Atrial natriuretic peptide: This peptide is primarily secreted by atrial stretch due to volume overload and left ventricular hypertrophy in the cirrhotics [105]. Echocardiographically enlarged atrium is an indirect evidence of high filling pressure. The raised level of ANP was found in cirrhotic patients with ascites [106-108].
B-type natriuretic peptide: BNP with its prohormone NTpro BNP is produced by the ventricles secondary to ischemia and ventricular pressure or volume overload. Secretion of NT-Pro hormone in ventricular overload counteracts Renin Angiotensin Aldosterone (RAAS) by natriuresis and water elimination [109]. Raised level of BNP and NT-pro BNP were found to be correlated with advanced cirrhosis, LV septal thickness, LV diameter in diastole, HR and QT interval. Raised level of BNP and NT-proBNP are correlated with end diastolic pressure (diastolic stretch). BNP and NT-proBNP have shown a well correlation with PCWP and E/e' ratios in cirrhotics even with normal LV function [18]. BNP is considered as an early marker of CCM [110]. NT-proBNP level >290pg/ml in a cirrhotic patient requires further cardiac evaluation [111].
Cardiac troponin: Troponins structural proteins are specific indicators of myocardial injury. Patients with alcoholic cirrhosis have shown elevated troponin I with an association to decrease cardiac output and ventricular mass without having any correlation to severity of cirrhosis [112]. Conversely troponin c levels well correlated with severity and mortality [113].
Adrenomedullin: Adrenomedullin hormone synthesized by cardiac tissue regulates natriuresis and vascular tone [114]. Its level is raised in cirrhotics both with and without CCM. Adrenomedullin is related to inotropism by activating cAMP which increases production of NO.
Novel cardiac biomarker: Galectin and copeptin [115,116] are newly investigated cardiac biomarker in cardiovascular disease. Galectin-3 level is found to be high in cirrhotics and been linked to myocardial fibrosis [117].
Echocardiography
Echocardiography is a noninvasive diagnostic modality for CCM. It is a screening tool for patients with liver cirrhosis. Earlier studies have shown various echocardiographic features as discussed earlier.
Cardiac doppler echocardiography: Diastolic dysfunction due to underlying ventricular relaxation delay is suggested by decrease and /or reversal of E/A ratio <1 [44,45], prolonged E wave deceleration time and isovolumetric relaxation time [44]. Echocardiographic assessment of Global Longitudinal Strains (GLS) has shown an effective parameter for detecting systolic and diastolic dysfunction [44].
Tissue doppler imaging: This is a more precise modality for detection of early diastolic mitral annulus velocity (e') which is considered specific for diastolic dysfunction. The criteria laid down by the American society of Echocardiography have been discussed earlier. Left atrial dysfunction can be assessed by speckle tracking.
Speckle tracking: Speckle tracking is an echocardiographic parameter which detects left ventricular myocardial strain in all three axes (radial, longitudinal and circumferential) [45]. Speckle tracking is being considered superior over 2D echocardiography for determination of systolic dysfunction.
ECG (Electrocardiogram)
Electrographic abnormalities in CCM including prolonged QT interval, atrial and ventricular premature contractions, bundle branch block and electromechanical dissociation have been discussed earlier.
Cardiac scintigraphy
There is limited data available on cardiac Scintigraphy in evaluation of CCM. Small increase in EF (a predictor of systole) after physiological stress can be revealed by Scintigraphy in cirrhotics when compared to non cirrhotic patients [118].
Magnetic Resonance Imaging (MRI)
Cardiac MRI is considered as a gold standard for accurate assessment of morphology of heart. It exactly defines the endocardial and pericardial borders.MRI with gadolinium contrast may determine EF, various volumes of cardiac chambers, myocardial fibrosis and edema [119]. MRI in cirrhotics have shown increase volume of left atrium, left ventricular hypertrophy and raised left ventricular volume at the end of diastole [44].
Management of CCM
To date there is no specific treatment available for CCM. CCM is treated like heart failure due to any other aetiology where sodium and water restriction, beta-blocker, diuretics and RAAS inhibitors are used [120]. Maintenance of sinus rhythm is mandatory as atrial fibrillation increases dyspnea and aggravates ascites in patients with cirrhosis.
β-blockers
Non selective agents (nodalol, propranalol) have shown a reduction in portal pressure, prevention of variceal bleeding and reduction in prolongation of QT interval. Carvedalol lies superior to traditional β blockers for reducing portal pressure. Nitrates association with β blocker has shown more reduction in preload, an important target for treatment of diastolic dysfunction [55]. Animal cirrhotic models have shown impairment of early diastole by β blockers [121].
Diuretics
Diuretics reduce preload in CCM with heart failure as they benefit in fluid retention, but their long term use may provoke electrolytes disturbance, hypovolemia and even HRS. Diuretics should be used cautiously in cirrhotic patients.
Cardiac glycosides and inotrops
Patients with CCM have shown down regulation of β-adrenoceptors inotrops like dobutamine and isoproterenol fails to improve diastolic dysfunction. Digitalis has also shown no improvement in systolic functions of patients with alcoholic cirrhosis as it can be dangerous [122]. It is not recommended for treatment of CCM.
Angiotensin Converting Enzyme Inhibitors (ACEl)
ACEI improves diastolic functions in cirrhotic patients by decreasing ventricular thickness and dilatation [123] and only be used in early cirrhosis (Child A). Major concerns with these drugs are hypotension and HRS. Currently there is no definite data available about use of ACEI in CCM.
Angiotensin-II receptor blockers
ACEI have not shown any clinical benefit in patients with cirrhosis [124]. Aldosterone antagonists have also shown reduction in ventricular wall thickness and end diastolic volume in early cirrhosis [125].
Steroids
Patients with CCM also have adrenal insufficiency which further compromises cardiac contractility. Earlier studies have demonstrated some improvement of cardiac function in this stress (hypoadrenalism) in CCM patients [126].
Liver transplantation
Liver transplantation is as an effective treatment modality of cirrhosis and CCM which has demonstrated improvement in cardiac dysfunction. Post transplant patients after 12 months have shown improvement in both diastolic and systolic function. Financial concerns, lack of availability, limited organ donors and the procedural risks like heart failure, myocardial infarction, tachyarrhythmia and cardiac death are major constraints of liver transplantation.
Conclusion
Cirrhosis of liver is a multi system disorder involving heart as CCM which is a characterized by systolic and diastolic dysfunction with electrophysiological abnormalities without having underlying known cardiac diseases. Major mechanisms for impaired systole explored by molecular insights include dysfunctional β-adrenoceptors, biochemical defect in cardiomyocyte membrane, channels impairment and enhanced activity of cardiac inhibitory neurohormones. Echocardiography with TDI precisely assessed the diastolic dysfunction (E/e' ratio). CCM well correlates with severity of cirrhosis where major clinical implications are circulatory deterioration and the development of HRS has no specific treatment but liver transplantation may restore cardiac functions.
References
- Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008; 371: 838-851.
- Møller S, Henriksen JH, Bendtsen F. Extrahepatic complications to cirrhosis and portal hypertension: haemodynamic and homeostatic aspects. World J Gastroenterol. 2014; 20: 15499-15517.
- Finucci G, Desideri A, Sacerdoti D, Sacerdoti D, Bolognesi M, Merkel C, et al. Left ventricular diastolic function in liver cirrhosis. Scand J Gastroenterol. 1996; 31: 279-284.
- Kowalski HJ, Abelmann WH. The cardiac output at rest in Laennec’s cirrhosis. J Clin Invest. 1953; 32: 1025-1033.
- Murray JF, Dawson AM, Sherlock S. Circulatory changes in chronic liver disease. Am J Med. 1958; 24: 358-367.
- Lunseth JH, Olmstead EG, Abboud F. A study of heart disease in one hundred eight hospitalized patients dying with portal cirrhosis. Arch Intern Med. 1958; 102: 405-413.
- Gould L, Shariff M, Zahir M, Di Lieto M. Cardiac hemodynamics in alcoholic patients with chronic liver disease and a presystolic gallop. J Clin Invest. 1969; 48: 860-868.
- Caramelo C, Fernandez-Munoz D, Santos JC, Blanchart A, Rodriguez-Puyol D, Lopez-Novoa JM, et al. Effect of volume expansion on hemodynamics, capillary permeability and renal function in conscious, cirrhotic rats. Hepatology. 1986; 6: 129-134.
- Pozzi M, Carugo S, Boari G, Pecci V, de Ceglia S, Maggiolini S, et al. Evidence of functional and structural cardiac abnormalities in cirrhotic patients with and without ascites. Hepatology. 1997; 26: 1131-1137.
- Bernardi M, Rubboli A, Trevisani F, Cancellieri C, Ligabue A, Baraldini M, et al. Reduced cardiovascular responsiveness to exercise-induced sympathoadrenergic stimulation in patients with cirrhosis. J Hepatol. 1991; 12: 207-216.
- Wong F, Girgrah N, Graba J, Allidina Y, Liu P, Blendis L. The cardiac response to exercise in cirrhosis. Gut. 2001; 49: 268-275.
- Valeriano V, Funaro S, Lionetti R, Riggio O, Pulcinelli G, Fiore P, et al. Modification of cardiac function in cirrhotic patients with and without ascites. Am J Gastroenterol. 2000; 95: 3200-3205.
- Zambruni A, Trevisani F, Caraceni P, Bernardi M. Cardiac electrophysiological abnormalities in patients with cirrhosis. J Hepatol. 2006; 44: 994-1002.
- Timoh T, Protano MA, Wagman G, Bloom M, Vittorio TJ. A perspective on cirrhotic cardiomyopathy. Transplant Proc. 2011; 43: 1649-1653.
- Donovan Baik SK, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Orphanet Journal of Rare Diseases. 2007; 2: 15.
- Donovan CL, Marcovitz PA, Punch JD, Bach DS, Brown KA, Lucey MR, et al. Two dimensional and dobutamine stress echocardiography in the prospective assessment of patients with end stage liver disease prior to orthotopic liver transplantation. Transplantation. 1996; 61: 1180-1188.
- Myers RP, Lee SS. Cirrhotic cardiomyopathy and liver transplantation. Liver Transplantation. 2000; 6: S44-S52.
- Ruiz-del-Arbol L, Achecar L, Serradilla R, RodrÍguez-Gandia MÁ, Rivero M, Garrido E, et al. Diastolic dysfunction is a predictor of poor outcomes in patients with cirrhosis, portal hypertension, and a normal creatinine. Hepatology. 2013; 58: 1732-1741.
- Naqvi IH, Mahmood K, Naeem M, Vaswani AS. The heart matters when the liver shatters! Cirrhotic cardiomyopathy: frequency, comparison, and correlation with severity of disease. Prz Gastroenterol. 2016; 11: 247-256.
- Arroyo V, Terra C, Ruiz-del-Arbol L, Rodés J, Benhamou JP, Blei AT, et al. Pathogenesis, Diagnosis and Treatment of Ascites in Cirrhosis. From Basic Science to Clinical Practice. Oxford: Blackwell Publishing Ltd. 2008: 666- 710.
- Lewis FW, Adair O, Rector WG. Arterial vasodilation is not the cause of increased cardiac output in cirrhosis. Gastroenterology. 1992; 102: 1024- 1029.
- Naqvi IH, Mahmood K, Talib A, Ubaid M, Mahmood A. Infections in Cirrhotics: Types, Microbiological Spectrum and Risk Factors-5-Year Cohort Study. Open Journal of Gastroenterology. 2014; 4: 105-117.
- Albillos A, de la Hera A, González M, Moya JL, Calleja JL, Monserrat J, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003; 37: 208-217.
- Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014; 60: 197-209.
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010; 140: 805-820.
- Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004; 95: 1140-1153.
- Benoit JN, Womack WA, Hernandez L, Granger DN. “Forward” and “backward” flow mechanisms of portal hypertension. Relative contributions in the rat model of portal vein stenosis. Gastroenterology. 1985; 89: 1092- 1096.
- Battarbee HD, Farrar GE, Spears RP. Responses to hypotension in conscious rats with chronic portal venous hypertension. Am J Physiol Gastrointest Liver Physiol. 1990; 259: 48-55.
- Zavecz JH, Bueno O, Maloney RE, O’Donnell JM, Roerig SC, Battarbee HD. Cardiac excitation-contraction coupling in the portal hypertensive rat. Am J Physiol Gastrointest Liver Physiol. 2000; 279: 28-39.
- Zardi EM, Dobrina A, Ambrosino G, Margiotta D, Polistina F, Afeltra A. New therapeutic approaches to liver fibrosis: a practicable route? Curr Med Chem. 2008; 15: 1628-1644.
- Sanyal AJ, Bosch J, Blei A, Arroyo V. Portal hypertension and its complications. Gastroenterology. 2008; 134: 1715-1728.
- Hendrickson H, Chatterjee S, Cao S, Morales Ruiz M, Sessa WC, Shah V. Influence of caveolin on constitutively activated recombinant eNOS: insights into eNOS dysfunction in BDL rat liver. Am J Physiol Gastrointest Liver Physiol. 2003; 285: 652-660.
- Bolognesi M, Sacerdoti D, Piva A, Di Pascoli M, Zampieri F, Quarta S, et al. Carbon monoxide-mediated activation of large conductance calciumactivated potassium channels contributes to mesenteric vasodilatation in cirrhotic rats. J Pharmacol Exp Ther. 2007; 321: 187-194.
- Wiese S, Hove JD, Bendtsen F, Moller S. Cirrhotic cardiomyopathy: pathogenesis and clinical relevance. Nat Rev Gastroenterol Hepatol. 2014; 11: 177-186.
- De BK, Majumdar D, Das D, Biswas PK, Mandal SK, Ray S, et al. Cardiac dysfunction in portal hypertension among patients with cirrhosis and noncirrhotic portal fibrosis. J Hepatol. 2003; 39: 315-319.
- Levy M, Wexler MJ. Renal sodium retention and ascites formation in dogs with experimental cirrhosis but without portal hypertension or increased splanchnic vascular capacity. J Lab Clin Med. 1978; 91: 520-536.
- Sarkar R, Meinberg EG, Stanley JC, Gordon D, Webb RC. Nitricoxide reversibly inhibits the migration of cultured vascular smoothmuscle cells. Circ Res. 1996; 78: 225-230.
- Laffi G, Barletta G, La Villa G, Del Bene R, Riccardi D, Ticali P, et al. Altered cardiovascular responsiveness to active tilting in nonalcoholic cirrhosis. Gastroenterology. 1997; 113: 891-898.
- Wong F, Liu P, Lilly L, Bomzon A, Blendis L. Role of cardiac structural and functional abnormalities in the pathogenesis of hyperdynamic circulation and renal sodium retention in cirrhosis. Clin Sci. 1999; 97: 259-267.
- Epstein M, Schneider N, Befeler B. Relationship of systemic and intrarenal hemodynamics in cirrhosis. J Lab Clin Med. 1977; 89: 1175-1187.
- Krag A, Bendtsen F, Mortensen C, Henriksen JH, Moller S. Effects of a single terlipressin administration on cardiac function and perfusion in cirrhosis. Eur J Gastroenterol Hepatol. 2010; 22: 1085-1092.
- Mikulic E, Munoz C, Puntoni LE, Lebrec D. Hemodynamic effects of dobutamine in patients with alcoholic cirrhosis. Clin Pharmacol Ther. 1983; 34: 56-59.
- Ramond MJ, Comoy E, Lebrec D. Alterations in isoprenaline sensitivity in patients with cirrhosis: evidence of abnormality of the sympathetic nervous activity. Br J Clin Pharmacol. 1986; 21: 191-196.
- Gassanov N, Caglayan E, Semmo N, Massenkeil G, Er F. Cirrhotic cardiomyopathy : A cardiologist’s perspective. World J Gastroenterol. 2014; 20: 15492-15498.
- Pall A, Czifra A, Vitalis Z, Papp M, Paragh G, Szabo Z. Pathophysiological and clinical approach to cirrhotic cardiomyopathy. J Gastrointestin Liver Dis. 2014; 23: 301-310.
- Glenn TK, Honar H, Liu H, ter Keurs HE, Lee SS. Role of cardiac myofilament proteins titin and collagen in the pathogenesis of diastolic dysfunction in cirrhotic rats. J Hepatol. 2011; 55: 1249-1255.
- Chayanupatkul M, Liangpunsakul S. Cirrhotic cardiomyopathy: review of pathophysiology and treatment. Hepatol Int. 2014; 8: 308-315.
- Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Sodium-induced cardiac aldosterone synthesis causes cardiachypertrophy. Endocrinology. 2000; 141: 1901-1904.
- Ma Z, Lee SS. Cirrhotic cardiomyopathy: getting to the heart of the matter. Hepatology. 1996; 24: 451-459.
- Wronski J, Fiedor P, Kwolczak M, Górnicka B. Retrospective analysis of liver cirrhosis influence on heart walls thickness. Pathol Res Pract. 2015; 211: 145-149.
- Yu HC, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, et al. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998; 98: 2621-2628.
- Moller S, Henriksen JH. Cardiovascular complications of cirrhosis. Gut. 2008; 57: 268-278.
- Henriksen JH, Schütten HJ, Bendtsen F, Warberg J. Circulating Atrial Natriuretic Peptide (ANP) and Central Blood Volume (CBV) in cirrhosis. Liver. 1986; 6: 361-368.
- Merli M, Calicchia A, Ruffa A, Pellicori P, Riggio O, Giusto M, et al. Cardiac dysfunction in cirrhosis is not associated with the severity of liver disease. Eur J Intern Med. 2013; 24: 172-176.
- Ruiz-del-Arbol L, Serradilla R. Cirrhotic cardiomyopathy. World J Gastroenterol. 2015; 21: 11502-11521.
- Karagiannakis DS, Vlachogiannakos J, Anastasiadis G, VafiadisZouboulis I, Ladas SD. Frequency and severity of cirrhotic cardiomyopathy and its possible relationship with bacterial endotoxemia. Dig Dis Sci. 2013; 58: 3029-3036.
- Moller S, Bernardi M. Interactions of the heart and the liver. Eur Heart J. 2013; 34: 2804-2811.
- Bernardi M, Calandra S, Colantoni A, Trevisani F, Raimondo ML, Sica G, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998; 27: 28-34.
- Zambruni A, Di Micoli A, Lubisco A, Domenicali M, Trevisani F, Bernardi M. QT interval correction in patients with cirrhosis. J Cardiovasc Electrophysiol. 2007; 18: 77-82.
- Mozos I. Arrhythmia risk in liver cirrhosis. World J Hepatol. 2015; 7: 662-672.
- Pall A, Czifra A, Vitális Z, Papp M, Paragh G, Szabó Z. Pathophysiological and clinical approach to cirrhotic cardiomyopathy. J Gastrointestin Liver Dis. 2014; 23: 301-310.
- Batchvarov V, Camm AJ. QT dispersion: measurement and interpretation. UpToDate. 2014.
- Mohamed R, Forsey PR, Davies MK, Neuberger JM. Effect of liver transplantation on QT interval prolongation and autonomic dysfunction in end-stage liver disease. Hepatology. 1996; 23: 1128-1134.
- Ytting H, Henriksen JH, Fuglsang S, Bendtsen F, Moller S. Prolonged Q-T(c) interval in mild portal hypertensive cirrhosis. J Hepatol. 2005; 43: 637-644.
- Genovesi S, Prata Pizzala DM, Pozzi M, Ratti L, Milanese M, Pieruzzi F, et al. QT interval prolongation and decreased heart rate variability in cirrhotic patients: relevance of hepatic venous pressure gradient and serum calcium. Clin Sci (Lond). 2009; 116: 851-859.
- Shin WJ, Kim YK, Song JG, Kim SH, Choi SS, Song JH, et al. Alterations in QT interval in patients undergoing living donor liver transplantation. Transplant Proc. 2011; 43: 170-173.
- Zambruni A, Trevisani F, Di Micoli A, Savelli F, Berzigotti A, Bracci E, et al. Effect of chronic beta-blockade on QT interval in patients with liver cirrhosis. J Hepatol. 2008; 48: 415-421.
- Zavecz JH, Bueno O, Maloney RE, O’Donnell JM, Roerig SC, Battarbee HD. Cardiac excitation-contraction coupling in the portal hypertensive rat. Am J Physiol Gastrointest Liver Physiol. 2000; 279: G28-G39.
- Henriksen JH, Fuglsang S, Bendtsen F, Christensen E, Møller S. Dyssynchronous electrical and mechanical systole in patients with cirrhosis. J Hepatol. 2002; 36: 513-520.
- Dahl EK, Møller S, Kjær A, Petersen CL, Bendtsen F, Krag A. Diastolic and autonomic dysfunction in early cirrhosis: a dobutamine stress study. Scand J Gastroenterol. 2014; 49: 362-372.
- Ruiz-del-Arbol L, Monescillo A, Jimenéz W, Garcia-Plaza A, Arroyo V, Rodés J. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology 1997; 113: 579-586.
- Hendrickse MT, Triger DR. Peripheral and cardiovascular autonomic impairment in chronic liver disease: prevalence and relation to hepatic function. J Hepatol. 1992; 16: 177-183.
- Hendrickse MT, Thuluvath PJ, Triger DR. Natural history of autonomic neuropathy in chronic liver disease. Lancet. 1992; 339: 1462-1464.
- Bajaj BK, Agarwal MP, Ram BK. Autonomic neuropathy in patients with hepatic cirrhosis. Postgrad Med J. 2003; 79: 408-411.
- Salerno F, Cazzaniga M. Autonomic dysfunction: often present but usually ignored in patients with liver disease. Liver Int. 2009; 29: 1451-1453.
- Frith J, Newton JL. Autonomic dysfunction in chronic liver disease. Liver Int. 2009; 29: 483-489.
- Ma Z, Meddings JB, Lee SS. Membrane physical properties mdetermine cardiac beta-adrenergic receptor function in cirrhotic rats. Am J Physiol. 1994; 267: G87-G93.
- Ma Z, Miyamoto A, Lee SS. Role of altered beta-adrenoceptor signal transduction in the pathogenesis of cirrhotic cardiomyopathy in rats. Gastroenterology. 1996; 110: 1191-1198.
- Gerbes AL, Remien J, Jüngst D, Sauerbruch T, Paumgartner G. Evidence for down-regulation of beta-2-adrenoceptors in cirrhotic patients with severe ascites. Lancet. 1986; 1: 1409-1411.
- Kakimoto H, Imai Y, Kawata S, Inada M, Ito T, Matsuzawa Y. Altered lipid composition and differential changes in activities of membrane-bound enzymes of erythrocytes in hepatic cirrhosis. Metabolism. 1995; 44: 825- 832.
- Ma Z, Lee SS, Meddings JB. Effects of altered cardiac membrane fluidity on beta-adrenergic receptor signalling in rats with cirrhotic cardiomyopathy. J Hepatol. 1997; 26: 904-912.
- Ceolotto G, Papparella I, Sticca A, Bova S, Cavalli M, Cargnelli G, et al. An abnormal gene expression of the beta-adrenergic system contributes to the pathogenesis of cardiomyopathy in cirrhotic rats. Hepatology. 2008; 48: 1913-1923.
- Ma Z, Meddings JB, Lee SS. Membrane physical properties determine cardiac beta-adrenergic receptor function in cirrhotic rats. Am J Physiol. 1994; 267: G87-G93.
- Jaue DN, Ma Z, Lee SS. Cardiac muscarinic receptor function in rats with cirrhotic cardiomyopathy. Hepatology. 1997; 25: 1361-1365.
- Keller M, Pignier C, Niggli E, Egger M. Mechanisms of Na+-Ca2+ exchange inhibition by amphiphiles in cardiac myocytes: importance of transbilayer movement. J Membr Biol. 2004; 198: 159-175.
- Mallat A, Teixeira-Clerc F, Lotersztajn S. Cannabinoid signaling and liver therapeutics. J Hepatol 2013; 59: 891-896.
- Caraceni P, Viola A, Piscitelli F, Giannone F, Berzigotti A, Cescon M, et al. Circulating and hepatic endocannabinoids and endocannabinoid-related molecules in patients with cirrhosis. Liver Int. 2010; 30: 816-825.
- Tam J, Liu J, Mukhopadhyay B, Cinar R, Godlewski G, Kunos G. Endocannabinoids in liver disease. Hepatology. 2011; 53: 346-355.
- Baldassarre M, Giannone FA, Napoli L, Tovoli A, Ricci CS, Tufoni M, et al. The endocannabinoid system in advanced liver cirrhosis: pathophysiological implication and future perspectives. Liver Int. 2013; 33: 1298-1308.
- Seddon M, Shah AM, Casadei B. Cardiomyocytes as effectors of nitric oxide signalling. Cardiovasc Res. 2007; 75: 315-326.
- Van Obbergh L, Vallieres Y, Blaise G. Cardiac modifications occurring in the ascitic rat with biliary cirrhosis are nitric oxide related. J Hepatol. 1996; 24: 747-752.
- Liu H, Ma Z, Lee SS. Contribution of nitric oxide to the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Gastroenterology. 2000; 118: 937-944.
- Mani AR, Ippolito S, Ollosson R, Moore KP. Nitration of cardiac proteins is associated with abnormal cardiac chronotropic responses in rats with biliary cirrhosis. Hepatology. 2006; 43: 847-856.
- Liu H, Song D, Lee SS. Role of heme oxygenase-carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. Am J Physiol Gastrointest Liver Physiol. 2001; 280: G68-G74.
- Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, et al. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial dependent apoptosis in ventricular myocytes. Circ Res. 2005; 97: 1009- 1017.
- Abbasi A, Joharimoqaddam A, Faramarzi N, Khosravi M, Jahanzad I, Dehpour AR. Opioid receptors blockade modulates apoptosis in a rat model of cirrhotic cardiomyopathy. Ann Med Health Sci Res. 2014; 4: 404-409.
- Nam SW, Liu H, Wong JZ, Feng AY, Chu G, Merchant N, et al. Cardiomyocyte apoptosis contributes to pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated mice. Clin Sci (Lond). 2014; 127: 519-526.
- Ren J, Zhang S, Kovacs A, Wang Y, Muslin AJ. Role of p38alpha MAPK in cardiac apoptosis and remodeling after myocardial infarction. J Mol Cell Cardiol. 2005; 38: 617-623.
- Gaasch WH, Battle WE, Oboler AA, Banas JS, Levine HJ. Left ventricular stress and compliance in man. With special reference to normalized ventricular function curves. Circulation. 1972; 45: 746-762.
- Bernardi M. Cirrhotic cardiomyopathy. Clin Liver Dis. 2013; 2: 99-101.
- Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008; 28: 59-69.
- Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Ginès P, Moreira V, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005; 42: 439-447.
- Krag A, Bendtsen F, Henriksen JH, Moller S. Low cardiac output predicts development of hepatorenal syndrome and survival in patients with cirrhosis and ascites. Gut. 2010; 59: 105-110.
- Moller S, Bendtsen F. Cirrhotic multiorgan syndrome. Dig Dis Sci. 2015; 60: 3209-3225.
- Rahman S, Mallett SV. Cirrhotic cardiomyopathy: implications for the perioperative management of liver transplant patients.World J Hepatol. 2015; 7: 507-520.
- Tsang TS, Barnes ME, Gersh BJ, Bailey KR, Seward JB. Left atrial volume as a morphophysiologic expression of left ventricular diastolic dysfunction and relation to cardiovascular risk burden. Am J Cardiol. 2002; 90: 1284- 1289.
- Gines P, Jiménez W, Arroyo V, Navasa M, López C, Tito L, et al. Atrial natriuretic factor in cirrhosis with ascites: plasma levels, cardiac release and splanchnic extraction. Hepatology. 1988; 8: 636-642.
- Salerno F, Badalamenti S, Moser P, Lorenzano E, Incerti P, Dioguardi N. Atrial natriuretic factor in cirrhotic patients with tense ascites. Effect of largevolume paracentesis. Gastroenterology. 1990; 98: 1063-1070.
- Licata A, Corrao S, Petta S, Genco C, Cardillo M, Calvaruso V, et al. NT pro BNP plasma level and atrial volume are linked to the severity of liver cirrhosis. PLoS One. 2013; 8: e68364.
- Wong F, Siu S, Liu P, Blendis LM. Brain natriuretic peptide: is it a predictor of cardiomyopathy in cirrhosis? Clin Sci (Lond). 2001; 101: 621-628.
- Raedle-Hurst TM, Welsch C, Forestier N, Kronenberger B, Hess G, Herrmann E, et al. Validity of N-terminal propeptide of the brain natriuretic peptide in predicting left ventricular diastolic dysfunction diagnosed by Tissue Doppler imaging in patients with chronic liver disease. Eur J Gastroenterol Hepatol. 2008; 20: 865-873.
- Pateron D, Beyne P, Laperche T, Logeard D, Lefilliatre P, Sogni P, et al. Elevated circulating cardiac troponin I in patients with cirrhosis. Hepatology. 1999; 29: 640-643.
- Wiese S, Mortensen C, Gotze JP, Christensen E, Andersen O, Bendtsen F, et al. Cardiac and proinflammatory markers predict prognosis in cirrhosis. Liver Int. 2014; 34: e19-e30.
- Welsh P, Hart C, Papacosta O, Preiss D, McConnachie A, Murray H, et al. Prediction of cardiovascular disease risk by cardiac biomarkers in 2 United Kingdom cohort studies: does utility depend on risk thresholds for treatment? Hypertension. 2016; 67: 309-315.
- Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004; 110: 3121-3128.
- Kimer N, Goetze JP, Bendtsen F, Moller S. New vasoactive peptides in cirrhosis: organ extraction and relation to the vasodilatory state. Eur J Clin Invest. 2014; 44: 441-452.
- Farr M, Schulze PC. Recent advances in the diagnosis and management of cirrhosis-associated cardiomyopathy in liver transplant candidates: advanced echo imaging, cardiac biomarkers, and advanced heart failure therapies. Clin Med Insights Cardiol. 2015; 8: 67-74.
- Rivas M. O acometimento cardiovascular na cirrose hepática : cardiomiopatia cirrótica. Rev SOCERJ. 1998; 11: 254-261.
- Lossnitzer D, Steen H, Zahn A, Lehrke S, Weiss C, Weiss KH, et al. Myocardial late gadolinium enhancement cardiovascular magnetic resonance in patients with cirrhosis. J Cardiovasc Reson. 2010; 12: 47.
- Bocchi EA, Braga FG, Ferreira SM, Rohde LE, Oliveira WA, Almeida DR, et al. Sociedade Brasileira de Cardiologia. III Diretriz brasileira de insuficiência cardÍaca crônica. Arq Bras Cardiol. 2009; 93: 3-70.
- Cheng CP, Igarashi Y, Little WC. Mechanism of augmented rate of left ventricular filling during exercise. Circ Res. 1992; 70: 9-19.
- Limas CJ, Guiha NH, Lekagul O, Cohn JN. Impaired left ventricular function in alcoholic cirrhosis with ascites. Ineffectiveness of ouabain. Circulation. 1974; 49: 754-760.
- Bos R, Mougenot N, Findji L, Mediani O, Vanhoutte PM, Lechat P. Inhibition of catecholamine-induced cardiac fibrosis by an aldosterone antagonist. J Cardiovasc Pharmacol. 2005; 45: 8-13.
- Vizzardi E, D’Aloia A, Giubbini R, Bordonali T, Bugatti S, Pezzali N, et al. Effect of spironolactone on left ventricular ejection fraction and volumes in patients with class I or II heart failure. Am J Cardiol. 2010; 106: 1292-1296.
- Pozzi M, Grassi G, Ratti L, Favini G, Dell’Oro R, Redaelli E, et al. Cardiac, neuroadrenergic, and portal hemodynamic effects of prolonged aldosterone blockade in postviral child A cirrhosis. Am J Gastroenterol. 2005; 100: 1110- 1116.
- Theocharidou E, Krag A, Bendtsen F, Moller S, Burroughs AK. Cardiac dysfunction in cirrhosis - does adrenal function play a role? A hypothesis. Liver Int. 2012; 32: 1327-1332.