
Case Report
Austin Gynecol Case Rep. 2016; 1(1): 1007.
McCune Albright Syndrome Affecting Only One of the Monozygotic Child of Twins: A Case Report
Ganie MA¹*, Raizada N¹, Kandaswamy DS² and Hammad-u-rehman¹
¹Department of Endocrinology, Metabolism and Diabetes, All India Institute of Medical Sciences, India
²Department of Radiodiagnosis, and Endocrinology, Metabolism and Diabetes, All India Institute of Medical Sciences, India
*Corresponding author: Mohd Ashraf Ganie, Department of Endocrinology, Metabolism and Diabetes, All India Institute of Medical Sciences, New Delhi, India
Received: July 28, 2016; Accepted: December 29, 2016; Published: December 30, 2016
Abstract
McCune Albright Syndrome (MAS) is a clinically rare condition that is characterized by a triad of poly/monostotic fibrous dysplasia, café-au-lait spots, and various endocrinopathies attributed to postzygotic somatic activating mutations of the guanine nucleotide-binding protein, alpha-stimulating activity polypeptide 1 (GNAS1). This peculiar pathogenesis makes the disorder intriguing to study in monozygotic twins. MAS has been infrequently reported in monozygotic twins and is actually a rarity. Hereby, we report a pair of monozygotic twins, one of whom was diagnosed with MAS while the twin sister of the affected girl, did not show any evidence of MAS on clinical, biochemical, hormonal and radiological evaluation. Although genetic analysis has not been performed, the mutation is a dominant one and the unaffected sister is unlikely to be carrying the mutated gene. Keeping in view the rarity of the disorder, this case reports needs to be documented for further understanding of this interesting disorder.
Keywords: McCune albright syndrome; Fibrous dysplasia; Café-au-lait spots; Endocrinopathies; Twins
Introduction
McCune Albright Syndrome (MAS) is a clinically rare condition that is characterized by a triad of poly/monostotic fibrous dysplasia, café-au-lait spots, and endocrinopathies. The disorder is known to be caused by postzygotic somatic activating mutations of the guanine nucleotide-binding protein, alpha-stimulating activity polypeptide 1 (GNAS1). This mutation leads to a mosaic of normal and mutant cells, and the extent and distribution of the mutant cells dictates the clinical profile of each patient. This peculiar pathogenesis makes it intriguing to study this disease in monozygotic twins. MAS has been infrequently reported in monozygotic twins. We report a pair of monozygotic twins, one of whom was diagnosed with MAS while the other had no stigmata of this syndrome.
Case Presentation
AB, the index patient and CD the sibling, were born out of a non-consanguineous marriage as a twin delivery. Soon after birth the parents had noted that AB had brownish patches on the right side of face, neck, chest and lower back. The girl had uneventful milestones and growth till she developed vaginal bleeding at the age of three years. She was diagnosed as gonadotropin independent precocious puberty but as the vaginal bleeding stopped spontaneously further medical attention was not sought. However, she again presented with development of breast and pubic hair at the age of 6 years. She was put on injectable gonadotropin releasing hormone agonists till the age of 11 years. After stopping the treatment, she attained menarche. At the age of four years, she had a fall while playing on level ground and complained of pain in her right thigh. Radiography revealed a fracture in the right femoral shaft for which closed reduction was performed and skin traction was given. The fracture united after a period of 3 months after which she was able to walk with a limp. She attained a height of 145 cm which was below the mid parental height (166 cm). The twin sibling had an uneventful childhood. She did not have the skin lesions unlike her sister. She started her breast development at the age of 10 years and attained menarche at 13 years of age. She achieved a height of 164 cm. There was no history of fracture or limp while walking.
On examination, the index patient had large cafe au lait spots on the right side of face, neck, upper chest and back (Figure 1). Her body mass index (weight in kilograms divided by the square of height in metres) was 19.0 kg/m2. She had facial dysmorphism and lurching gait. Scoliosis of lumbar spine was noted. She had a diffuse goitre (WHO grade II). However, there were no clinical features to suggest thyrotoxicosis. There was no evidence of acral enlargement or other features of growth hormone excess. There were no striae, skin thinning or proximal myopathy.

Figure 1: Showing front view and lateral profile of the patient demonstrating
Caif au laite spot.
Her hemogram, liver and kidney function tests were unremarkable. The serum total calcium was 9.4 mg/dl (normal range 8.5-10.3 mg/ dl) with a serum phosphate of 4.3 mg/dl (normal range 2.5-5.5 mg/ dl) and serum alkaline phosphtase of 184 IU/L (normal range 44-180 IU/L). Her investigations revealed normal free T4 levels (1.50 ng/dl, normal range 0.93-1.70 ng/dl) with suppressed TSH (0.1microIU/ ml, normal range 0.27-4.2 microIU/ml). Radioiodine uptake was increased (10% at 2 hours and 45% at 24 hours) while technetium 99 thyroid scan revealed a hot nodule in left lobe of thyroid. Her serum IGF -1 levels were also normal (330 micrograms/l, normal range 180-780 micrograms/l). Technetium 99 bone scintigraphy revealed uptake at several sites including calvarium, mandible, multiple ribs, left scapula, left humeral shaft, multiple dorsolumbar vertebrae, pelvis, bilateral femur and tibia which was consistent with polyostotic fibrous dysplasia. Skeletal survey showed lesions consistent with fibrous dysplasia at these sites (Figure 2a and 2b). There was no objective hearing loss on pure tone audiometry and visual evoked response testing was normal.
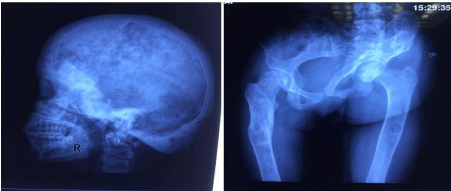
Figure 2: Showing multiple lytic lesions and ground glass appearance of
radiographs of (a) Skull lateral view (b) Pelvis AP view.
The twin sister was evaluated for features of MAS. She did not have café au lait macules or skeletal deformities at the time of evaluation. There were no clinical features to suggest Cushing’s syndrome, thyroid hormone or growth hormone excess. Her hemogram, liver and kidney function tests were normal. Her skeletal survey, bone scan and hormonal profile were normal. Her investigations revealed normal free T4 levels (1.20 ng/dl, normal range 0.93-1.70 ng/dl) with normal TSH (3.5 microIU/ml, normal range 0.27-4.2 microIU/ml). Her serum IGF-1 levels were also normal (400 micrograms/l, normal range 180-780 micrograms/l).
Discussion
The original description of MAS was given by DJ McCune in 1936 where he described skeletal lesions (fibrous dysplasia), café au lait macules, sexual precocity and hyperthyroidism [1]. The pathogenesis of the disorder was proposed by Happle who observed that the cutaneous hyperpigmentation in MAS follows the developmental lines of Blaschko. He proposed that the underlying genetic abnormality is a dominant somatic mutation occurring early in development leading to a mosaic with a widespread distribution of mutant cells [2]. He proposed that the mutation would be lethal if present in the germline because MAS is almost never inherited. The disorder is known to occur as a result of a postzygotic somatic activating mutation of GNAS (encoding the cAMP pathway-associated G-protein, Gsα). Depending upon the time of occurrence and the site of this acquired mutation, various organs of the body will contain a different proportion of the mutant and normal cells. The clinical manifestations will be therefore the outcome of the proportion of these cell populations. Later on, the mutation in MAS was described to occur in the GNAS gene at 20q13, which encode constitutively activated forms of Gs alpha. The mutated Gs alpha subunit undergoes prolonged activation, even in the absence of any signalling. This is because of abnormality in the intrinsic GTPase mechanism which is critical for inactivation of the Gs alpha subunit. This excess cyclic AMP thus generated leads to downstream events manifesting as the various clinical features of MAS, including bone marrow stromal cell proliferation (fibrous dysplasia), melanocyte activation (café au lait spots) [3], pituitary tumorigenesis and Endocrinopathies (due to enhanced downstream activity of cAMP dependent hormones like GH releasing hormone (GHRH), TSH, ACTH and gonadotropins).
Fibrous dysplasia can involve any part or combination of the craniofacial, axial, and/or appendicular skeleton (monostotic lesion or polyostotic disease) [4]. Scoliosis, facial deformity, pathological fractures, loss of vision and hearing (cranial nerve compression) can occur due to fibrous dysplasia. Café-au-lait skin macules are usually the first manifestation of the disease, apparent at or shortly after birth. Various endocrinopathies include recurrent ovarian cysts among girls and Leydig cell hyperactivity in boys causing gonadotropinindependent precocious puberty [5]. Poorly treated gonadotropnindependent precocious puberty can later manifest as true precocious puberty. Neonatal hypercortisolism can also be seen [6]. The affected individuals can present with or without thyrotoxicosis thyroid nodules. Acromegaly or gigantism can be due to growth hormone excess either because of GHRH hyperactivity or due to GH secreting pituitary adenoma [7]. Besides subjects can have FGF-23 mediated phosphate wasting that can occur especially with polyostotic fibrous dysplasia with a large burden of skeletal disease.
The cause of these mutations is unclear. Further at what time after fertilization does the mutation occur is also not well understood? Monozygotic twins, as our duo seems to be, might provide a novel outlook to the pathogenesis of this disorder. As MAS is relatively a rare disease, MAS in monozygotic twins is a rarity. Our search of literature revealed only three case reports of McCune Albright occurring in one of monozygotic twins. Lemli [8] reported monozygotic twins with and without McCune Albright syndrome in 1977. One of the twins had sexual precocity and growth hormone excess. Endo et al. [9] reported a girl, one of Monozygotic (MZ) twins, with precocious puberty, café-au-lait spots and polyostotic fibrous dysplasia. The co-twin had an advanced bone-age and a small café-au-lait spot without any evidence of precocity or fibrous dysplasia suggesting a forme fruste of the disorder. The authors proposed a 2-hit mutation hypothesis: an inherited dominant mutation followed by a second mutation occuring in somatic cell in the early somatic division. The resulting mosaicism could therefore explain the discrepancy between the manifestations among the co- twins. Our co-twin had no historical, clinical, radiological, biochemical and hormonal abnormalities even though phenotypically they were identical.
Peleg et al. [10] tried to identify differences in the GNAS1 gene in DNA extracted from peripheral blood cells and mRNA extracted from lymphoblastoid cells attained from monozygotic twin sisters, one of whom had fibrous dysplasia and growth hormone excess. They could not identify any mutations in the DNA of both the sisters and nor was the mRNA quantity different in the lymphoblastoid cells. They proposed that the mutation occurred after separation from the healthy twin and this mutation may not have been distributed to all tissues.
In our case, the twin sister of the affected girl did not show any evidence of MAS on clinical, biochemical, hormonal and radiological evaluation. Although genetic analysis has not been performed, the mutation is a dominant one and the unaffected sister is unlikely to be carrying the mutated gene. We can speculate that the mutation has occurred after the zygote divided into two; why one of the resultant zygotes acquired the mutation is a question which has no definitive answer at present. Keeping in view the rarity of this disorder, this case report needs to be documented. The understanding of this interesting disorder will improve, if the answers to the above questions can be found.
References
- McCune DJ. Osteitisfibrosacystica: the case of a nine year old girl who also exhibits precocious puberty, multiple pigmentation of the skin and hyperthyroidism. Am J Dis Child. 1936; 52: 743-747.
- Happle R. The McCune-Albright syndrome: A lethal genesurviving by mosaicism. Clin Genet. 1986; 29: 321-324.
- Kim I, Kim ER, Nam HJ, Chin MO, Moon YH, Oh MR, et al. Activating mutation of Gs in McCune-Albright syndrome causes skin pigmentation by tyrosinase gene activation on affected melanocytes. Horm Res. 1999; 52: 235-240.
- Marie PJ, Lomri A, Chanson P, de Pollak C. Increased proliferation of osteoblastic cells expressing the activating Gs alpha mutation in monostotic and polyostotic fibrous dysplasia. Am J Pathol. 1997; 150: 1059-1069.
- Roman R, Johnson MC, Codner E, Boric MA, Avila A, Cassorla F. Activating GNAS1 gene mutations in patients with premature thelarche. J Pediatr. 2004; 145: 218-222.
- Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MA, Zerbini MC, et al. Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metab. 2003; 88: 2147-2151.
- Wortham JT, Hanblem EC. Polyostotic fibrous dysplasia with sexual precocity and pigmentation (Albright’s syndrome) and associated non neoplastic acromegaly. J Clin Endocrinol Metab. 1952; 12: 975-976.
- Lemli L. Fibrous dysplasia of bone. Report of female monozygotic twins with and without the McCune-Albright syndrome. J Pediatr. 1977; 91: 947-949.
- Endo M, Yamada Y, Matsuura N, Niikawa N. Monozygotic twins discordant for the major signs of McCune Albright syndrome. Am J Med Genet. 1991; 41: 216-220.
- Peleg R, Luba A, Eliakim A, Israeli-Shani L, Manor E, Birk R, et al. McCune- Albright syndrome in a discordant monozygotic twin. Isr Med Assoc J. 2009; 11: 343-347.