
Review Article
Austin Gynecol Case Rep. 2017; 2(1): 1008.
Mechanisms of Luteinising Hormone Regulation in Female Steroidogenesis
Cadagan D*
Department of Sciences, Staffordshire University, England
*Corresponding author: Cadagan D, Lecturer in Biomedicine, Senior Biomedical Researcher, Department of Sciences, Staffordshire University, England
Received: June 10, 2016; Accepted: January 10, 2017; Published: January 12, 2017
Abstract
Understanding the mechanisms of steroidogenic regulation in female fertility is fundamental to determining dysfunction. Research has revealed a strong relationship between fertility and metabolic disturbances. With many endocrine signals involved in activation of steroidogenic cascades, it has become necessary to map the complex cross-talk between these regulatory systems and this review looks to establish this based on current understanding. Luteinising hormone is a vital endocrine hormone that is already known to signal via these pathways, although its involvement in metabolic disturbance is less understood. Studies investigating activation of the LH receptor in key steroidogenic cells, ovarian theca and granulosa, have highlighted overlaps in important signalling cascades including PKC/MAPK/PKA and PI3K. Here, we review LH and its signalling cross-talk in key steroidogenic pathways.
Keywords: Luteinising hormone (LH); Lutropin; Steroidogenesis; Gonadotropin; Ovulation; LH Receptor; Cyclic adenomonophosphate (cAMP); Diacylglycerol (DAG); Protein kinase C pathway; MAPK pathway
Introduction
The pituitary gland is responsible for secretion of various endocrine signalling hormones with the anterior pituitary producing somatotropins, thyrotropins, corticotropins, lactotropins and gonadotropins. Dysfunction of the pituitary gland can therefore have systemic effects as a result of signalling errors and conditions such as hyper and hypopituitarism. These dysfunctions have been linked to the pathophysiology of conditions such as diabetes and the metabolic syndrome. There has been a rapid increase in Diabetes Mellitus (DM) over the past decades resulting in it becoming one of the most prevalent endocrine disorders, effecting ~135 million people. Therefore, it is vital to understand the mechanisms behind the condition [1,2].
Many studies have examined the effect of diabetes on gonadotropin production with an aim of determining a relationship between impaired fertility and menstrual disturbances [3-5]. In doing so, the importance of Luteinising Hormone (LH) in pituitary regulation has been highlighted, as well as its sensitivity to metabolic changes [1]. Although reduced gonadotropin response may play only a partial role in steroidogenic function it is important to understand the mechanism and pathways involved in order to fully understand the overall dysfunction.
This review will examine our current understanding of the effects of LH on steroidogenic cells to allow future mapping of possible regulatory pathways that may be seen as targets for dysfunction in metabolic diseases.
Luteinising Hormone and Steroidogenesis
The gonadotropins are a family of proteins consisting of LH, Follicular Stimulating Hormone (FSH) and the Chorionic Gonadotropins (CG). These hormones are vital in the control and regulation of reproductive function in males and females. LH and FSH are secreted by the anterior pituitary gland, whereas CG is produced by a component of the fertilised egg called the Syncytiotrophoblast. LH, also known as lutropin, stimulates ovulation and development of the corpus luteum females. Structurally LH is a heterodimeric glycoprotein similar in structure to FSH, thyroid stimulating hormone and Human Chorionic Gonadotropin (hCG). All four proteins share a common alpha subunit, with the beta subunits conferring biological specificity [6,7].
In females, FSH action on ovarian granulosa cells initiates follicular growth. This stimulates a rise in oestrogen and LH receptor expression. Eventual positive feedback to the hypothalamus occurs, resulting in the release of LH over a 24-48hr period. The ensuing LH surge triggers ovulation, releasing the egg and initiating the production of the corpus luteum from the remnants of the follicle. The corpus luteum secretes progesterone for endometrial preparation and the possibility of implantation. LH is maintained for up to two weeks allowing for steroidogenesis along with support via hCG action. In the case of pregnancy LH supports theca cells which produce androgens necessary for estradiol synthesis and cell proliferation [3,7].
LH Receptor
Luteinising hormone / chorionic gonadotropin activates multiple signal transduction systems. The luteinising hormone receptor is part of the Glycoprotein Hormone Receptor Family (GpHRs) which also includes Follicular Stimulating Hormone Receptor (FSHR) and Thyroid Stimulating Hormone Receptor (TSHR). The LH receptor differs from these through its ability to bind two glycoprotein hormones, LH and hCG. These receptors are responsible for regulation of reproductive and metabolic processes with LHR activation leading to androgen synthesis and ovulation. The GpHRs contain two major domains of approximately the same size. Firstly, a large, glycosylated N-terminal Ectodomain (ECD) containing Leucine-Rich Repeats (LRRs) capped by cys-rich regions, the latter forming a portion of a hinge region. Secondly, a Trans-Membrane domain (TM) with seven membrane spanning helices, three extracellular loops (ecls), three intracellular loops (icls) and a short icl 4, an eighth cytoplasmic helix parallel to the plasma membrane, and a cytoplasmic tail [3]. The ECD and TM domains have important and distinct functional roles, namely hormone binding and signal transduction, respectively [8,9].
Most of the sequential steps involved after hormone binding to the ECD until G protein activation on the inner face of the plasma membrane remain poorly understood. In many experimental systems, LH or hCG binding to LHR results in activation of both protein kinase A and protein kinase C. At relatively low concentrations of LH and hCG, Gs appears to be the preferred signalling pathway, resulting in a rapid increase in the intracellular concentration of cAMP [10].
Cyclic Adenomonophosphate
cAMP is derived from adenosine triphosphate and is utilised within intracellular signal transductions as a second messenger, transferring the effects of hormones incapable of permeating cellular membranes. Typically, it allows activation of protein kinases and is also involved in the release of intracellular calcium from the endoplastic reticulum.
cAMP synthesis occurs through adenlyl cyclase action on ATP located on the inner plasma membrane and decomposition occurs through phosphodiesterase action on cAMP. This action occurs through stimulatory G-protein (Gs)-coupled receptor activation and is inhibited by agonists of adenylyl cyclase inhibitory G (Gi)-proteincoupled receptors.
Adenylate cyclase can be activated or inhibited by G proteins, which are coupled to membrane receptors and thus can respond to hormonal or other stimuli. Following activation cAMP acts as a second messenger by interacting with and regulating other proteins such as protein kinase A and cyclic nucleotide-gated ion channels [11,12].
Studies in pre-pubertal female rats have shown that in conditions such as uraemia can lead to reduced gonadtropin - cAMP sensitivity. This condition is related to metabolic disturbance and has direct associations to diabetes mellitus and diabetes induced cardiac failure as well as chronic kidney failure [13]. Furthermore, female uremic patients have shown menstrual irregularities and cases of delayed puberty possibly associated with gonadotropin target response. However, research in uremic premenopausal women has also shown hormonal disturbances including an absence in pre-ovulatory peaks of LH and estradiol concentrations. The source of these irregularities may therefore originate within the hypothalamus via impaired production of GnRH or in the anterior pituitary rather than within the cellular targets [14].
Diacylglycerol / Protein Kinase C Pathway
Diacylglycerol (DAG) is a key second messenger that regulates important cellular responses including proliferation, apoptosis, immune response and differentiation. This occurs via its target protein Protein Kinase C (PKC). Activation is linked to the LH receptor as a hepta-membrane spanning protein that couples stimulatory binding proteins Gs and Gq to allow adenylate cyclise signalling and increases in cAMP. This subsequently activates cAMP dependent PKA [11,12].
LH receptor activation increases intracellular calcium as a result of Phospholipase C activation (PLC) [5,15]. Once activated PLC allows cleavage of phosphatidylinsitol, 4,5-biphospate into inositol- 1,4,5-triphosphate (IP3) and Diacyglycerol (DAG). IP3 then binds to intracellular calcium channels on the endoplasmic reticulum resulting in increased cytosoliccalcium levels. DAG binds to Protein Kinase C (PKC) allowing co-activation (Figure 1) [16,17]. PKC requires calcium, phosphatitylserine and DAG for activation. These regulators allow a conformational change revealing the substrate binding site [18].
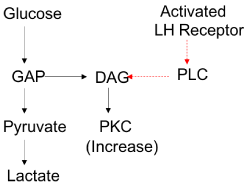
Figure 1: PKC pathway showing crosstalk between glucose and LH
activation.
Mitogen Activating Protein Kinase (MAPK) activation has been found to be independent of PKC activation in ovarian granulosa cells. This suggests an independent pathway that maybe regulated by LH alone. Furthermore, PKC has been seen to be partially activated by glucose and GAP in the absence of LH receptor activation in granulosa cells which may allow for pathway cross talk [19].
MAPK Pathway
Ras proteins are ubiqtous GTPases located on the inner surface of plasma membranes. They communicate receptor-mediated signal transduction pathways, transmitting extracellular signals that promote growth, proliferation, and differentiation. The signalling cascade starts from the plasma membrane where the growth factor binds to its enzyme-linked receptor. Receptor-linked tyrosine kinases such as the Epidermal Growth Factor Receptor (EGFR) are activated by extracellular ligands and binding of EGF’s. This leads to phosphorylation of the intracellular parts of the receptors. Activating Guanine Exchange Factors (GEFs), such as SOS and GEFs allow the exchange of GDP to GTP activating Ras [16]. The major downstream target of Ras-GTP is Mitogen-Activated Protein Kinase (MAPK), but also activates phosphatidylinositol 3-kinase (PI3-kinase), Ras-related guanine nucleotide dissociation stimulator (RalGDS). Activation of MAPK occurs through specific phosphorylation of Raf, which is activated on the plasma membrane by Ras-GTP. Raf phosphorylates Mitogen-Activated Kinase 1/2 (MEK1/2 kinase), in turn activating the extracellular regulated kinase 1/2 (ERK1/2 kinase or p44/42 MAPK) by phosphorylation.
The second class of enzymes in the ERK cascade are the Mitogen- Activated Protein Kinases (MEKs). These phosphorylate threonine andtyrosine residues. MEK1 and MEK2 are involved in ERK signalling and activated via there only substrate, Raf kinases. Inactivation of the ERKs can occur through de-phosphorylation via serine threonine or tyrosine phosphatises [20,21].
ERK1/2 kinase phosphorylates a variety of downstream targets, which results in changes in gene expression and the catalytic activities of enzymes and makes them important targets in steroidogenic activity [22].
A recent study showed that both MAPK kinase (MEK1/2) phosphorylation and extracellular signal related kinase (ERK1/2) phosphorylation were reduced by 50% in Polycystic Ovarian Syndrome (PCOS) steroidogenic cells compared with that in normal cells. A reduction in MEK1/2 and ERK1/2 phosphorylation is associated with increased theca CYP17 gene expression and subsequent androgen biosynthesis [23].
Treatment of ovarian steroidogenic theca cells with the MEK inhibitor PD98059 has been shown to augment theca CYP17 gene expression and androgen biosynthesis [23]. The extent to which the other components of the MAPK pathway contribute to overall androgen biosynthesis requires further examination.
Although LH has been reported to activate the cAMP/PKA pathway and the ERK/MAPK pathway in theca cells, whether and how LH stimulates the PI3K/Akt cascade in theca cells remains unclear. Recent studies have shown LH stimulates Akt phosphorylation in both cultured bovine and human theca cells, and that activation of PI3K/Akt is involved in CYP17A1 mRNA expression and androgen production in theca cells thus supporting LH activation of PI3K [24- 26]. H89, a potent and selective inhibitor of PKA, did not affect LHmediated changes in phospho-Akt, indicating that a pathway distinct from that of the PKA is involved in LH-induced Akt phosphorylation in theca cells.
In contrast to PKA inhibitor, the MEK inhibitor (U0126) blocked LH-mediated Akt phosphorylation and androgen production in theca cells. This suggests that LH stimulates CYP17A1 mRNA expression and androgen production in theca cells via activation of the PI3K/Akt pathway and with the involvement of the MAPK pathway.
While the precise mechanism for the activation of PI3K pathway by LH in theca cells is not known, we propose LH-induced phospho- Akt up-regulation may involve MAPK-mediated down-regulation of phosphatase and tensin homologue (PTEN) a tumour suppressor which negatively regulates Akt phosphorylation. In this context, it has been shown that PI3Kis required for estradiol-stimulated hepatic cell growth and that the MAPK pathway reduces the level of PTEN, allowing estradiol-induced phosphorylation of Akt [27]. Whether this indeed is the case in the theca cells awaits further investigation.
Discussion
Clarification of the LH-mediated intracellular signalling events is essential along with consideration for cross talk and compensatory mechanisms between pathways. This will allow for better understanding of not only ovarian physiology, but also of the pathophysiology of steroidogenic dysfunction.
Studies in ovarian steroidogenic granulosa cells have shown elevated cAMP and activated ERK. PKA forms inactive RAP1 inhibiting RAF1 and preventing MEK phosphorylation of ERK and subsequent androgen synthesis [28]. Based on these findings increased cAMP may allow phosphorylation of cAMP-responsive guanine nucleotide exchange factors (Epac1 and Epac2). Upon binding of cAMP, Epac1/2 may rapidly activate Rap1, which subsequently promotes activation of B-raf and the rest of the ERK cascade [29].
Direct activation by cAMP, RAP1 and subsequent BRAF activation would lead to MEK phosphorylation (LH mediated androgen synthesis). Inhibition of PKA would prevent RAP1 formation and eventual RAF1 inhibition would return due to reduced RAP1 formation (Figure 2). This is supported by studies showing LH mediated ERK phosphorylation only occurring within 24hrs of LH exposure during PKA inhibition [30].
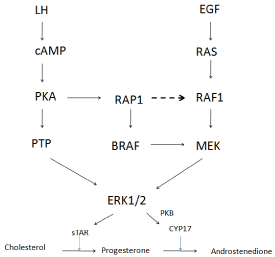
Figure 2: Proposed signal cascade / crosstalk in steroidogenesis, following
LH and EGF activation.
With the importance of steroidogenic regulation in fertility and our understanding of the relationship this may have with metabolic dysfunction, it is important to understand cross talk within regulatory pathways and the involvement of hormonal regulators. LH continues to show a vital role in multiple signalling cascades and its specific involvement requires further examination.
References
- Distiller LA, Sagel J, Morley JE, Joffe BI, Seftel HC. Pituitary Responsiveness to Luteinizing Hormone-Releasing Hormone in Insulin-Dependent Diabetes- Mellitus. Diabetes. 1975; 24: 378-380.
- Doi SAR, Towers PA, Scott CJ, Al-Shoumer KAS. PCOS: an ovarian disorder that leads to dysregulation in the hypothalamic-pituitary-adrenal axis? Eur J Obstet Gyn R B. 2005; 118: 4-16.
- Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002; 23: 141-174.
- Ascoli M. Molecular basis of the regulation of the lutropin/choriogonadotropin receptor. Biochem Soc T. 1997; 25: 1021-1026.
- Mestman JH, Schmidt sarosi C: Diabetes-Mellitus and Fertility-Control - Contraception Management Issues. American Journal of Obstetrics and Gynecology. 1993; 168: 2012-2020.
- Schroeder F, Frolov A, Schoer J, Gallegos A, Atshaves B, Stolowich N, et al. Intracellular Cholesterol Trafficking. 1998; 213-234.
- Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991; 124: 43-101.
- Dufau ML, Morris CHT, Hu ZZ, Geng Y, Xie X, Zhuang L, et al. The Luteinizing-Hormone Receptor. Front Endocrinol. 1994, 5: 51-60.
- Schroeder F, Frolov A, Schoer JK, Gallegos AM, Atshaves BP, Stolowich NJ, et al. Intracellular sterol binding proteins: Cholesterol transport and membrane domains. Intracellular Cholesterol Trafficking. 1998; 213-234.
- Gilchrist RL, Ryu KS, Ji I, Ji TH. The luteinizing hormone/chorionic gonadotropin receptor has distinct transmembrane conductors for cAMP and inositol phosphate signals. J Biol Chem. 1996; 271: 19283-19287.
- Hsueh AJW, Adashi EY, Jones PBC, Welsh TH. Hormonal Regulation of the Differentiation of Cultured Ovarian Granulosa Cells. Endocrine Reviews. 1984; 5: 76-127.
- Richards JS. Maturation of Ovarian Follicles - Actions and Interactions of Pituitary and Ovarian Hormones on Follicular Cell-Differentiation. Physiological Reviews. 1980; 60: 51-89.
- Kreusser W, Mader H, Haag WD, Ritz E. Diminished Response of Ovarian Camp to Luteinizing-Hormone in Experimental Uremia. Kidney International. 1982; 22: 272-279.
- Ginsburg ES, Owen WF. Reproductive Endocrinology and Pregnancy in Women on Hemodialysis. Semin Dialysis. 1993; 6: 105-116.
- Salvador LM, Maizels E, Hales DB, Miyamoto E, Yamamoto H, Hunzicker- Dunn M. Acute signaling by the LH receptor is independent of protein kinase C activation. Endocrinology. 2002; 143: 2986-2994.
- Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997; 9: 161-167.
- Zhu X, Gilbert S, Birnbaumer M, Birnbaumer L. Dual signaling potential is common among Gs-coupled receptors and dependent on receptor density. Mol Pharmacol. 1994; 46: 460-469.
- Iyengar R, Birnbaumer L. Roles of G proteins and G protein subunits in signal transduction. Lymphokine Res. 1990; 9: 533-537.
- Geffner M, Kaplan S, Bersch N, Golde D, Landaw E, Chang R. Persistence of insulin resistance in polycystic ovarian disease after inhibition of ovarian steroid secretion. Fertil Steril. 1986; 45: 327-333.
- Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000; 14: 6-16.
- Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000; 12: 186-192.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. The Molecular Biology of the Cell. Garland Science. New York. 2002.
- Nelson-Degrave VL, Wickenheisser JK, Hendricks KL, Asano T, Fujishiro M, Legro RS, et al. Alterations in mitogen-activated protein kinase kinase and extracellular regulated kinase signaling in theca cells contribute to excessive androgen production in polycystic ovary syndrome. Mol Endocrinol. 2005; 19: 379-390.
- Fukuda S, Orisaka M, Tajima K, Hattori K, Kotsuji F: Luteinizing hormoneinduced Akt phosphorylation and androgen production are modulated by MAP Kinase in bovine theca cells. J Ovarian Res. 2009; 2: 17.
- Cadagan D, Khan R, Amer S. Thecal cell sensitivity to luteinizing hormone and insulin in polycystic ovarian syndrome. Reprod Biol. 2016; 16: 53-60.
- Fukuda S, Orisaka M, Tajima K, Kotsuji F. Luteinizing Hormone Stimulates CYP17A1 mRNA Expression and Androgen Production in Bovine Theca Cells via Activation of the PI3K/Akt Pathway. Biology of Reproduction. 2009; 81.
- Marino M, Acconcia F, Trentalance A. Biphasic estradiol-induced AKT phosphorylation is modulated by PTEN via MAP kinase in HepG2 cells. Mol Biol Cell. 2003; 14: 2583-2591.
- Tajima K, Yoshii K, Fukuda S, Orisaka M, Miyamoto K, Amsterdam A, et al. Luteinizing hormone-induced extracellular-signal regulated kinase activation differently modulates progesterone and androstenedione production in bovine theca cells. Endocrinology. 2005; 146: 2903-2910.
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998; 396: 474-477.
- Seger R, Hanoch T, Rosenberg R, Dantes A, Merz WE, Strauss JF 3rd, et al. The ERK signaling cascade inhibits gonadotropin-stimulated steroidogenesis. J Biol Chem. 2001; 276: 13957-13964.