
Case Report
Austin Gynecol Case Rep. 2017; 2(1): 1010.
Identification of a Novel Variant in TCTN2 using Clinical Exome Sequencing in a South Indian Family with Recurrent Meckel-Gruber Syndrome
Prasoona KR, Sunitha T, Srinadh B and Jyothy A*
Institute of Genetics and Hospital for Genetic Diseases, Osmania University, India
*Corresponding author: Jyothy A, Former Professor, Institute of Genetics and Hospital for Genetic Diseases, Osmania University, Begumpet, Hyderabad, Telangana State, India
Received: October 29, 2016; Accepted: January 20, 2017; Published: January 23, 2017
Abstract
Background: Meckel-Gruber Syndrome (MGS) is a rare autosomal recessive lethal ciliopathy characterized by a wide variety of systemic malformations. The most common features are occipital encephalocele, multi cystic dysplasia of kidney, cystic and fibrotic changes of liver, and polydactyly.
Methods: In this report, we present a 25 year-old-pregnant woman of G4P1L0D1A2 with history of MGS in her three pregnancies. In the present fourth pregnancy also, second-trimester ultrasound scan showed features of MGS. The patient was counselled regarding the lethal outcome of MGS and the family opted for termination of pregnancy. Amniocentesis was done and prenatal clinical exome sequencing of the foetus was performed using Illumina NGS platform. Ten genes (B9D1, B9D2, CC2D2A, CEP290, MKS1, NPH3, RPGRIP1L, TCTN2, TMEM216, TMEM67) associated with MGS were analysed in addition to pathogenic and likely pathogenic variants present in the exome panel.
Results: Clinical exome sequencing revealed a novel heterozygous missense substitution (c.1949C>T, p.Pro650Leu) in exon 17 of the TCTN2 gene. In silico missense prediction tools suggest that this variant alters a conserved residue and is predicted to be damaging the protein function. No other known pathogenic or likely pathogenic variants were identified.
Conclusion: By applying clinical exome sequencing, we could identify a novel variant in the TCTN2 gene in a family with recurrent MGS that may influence the disease risk.
Keywords: Meckel-Gruber Syndrome (MGS); Occipital encephalocele; Polydactyly; TCTN2 gene; Clinical exome sequencing
Case Presentation
A 25-year-old pregnant woman with G4P1L0D1A2 was referred for antenatal scan and genetic counselling at 14 weeks of gestation to Institute of Genetics and Hospital for Genetic Diseases, Osmania University. Her last menstrual period was confirmed by ultrasound and the pregnancy was conceived naturally. Family history revealed first degree consanguineous marriage (Figure 1). Clinical history of the couple was normal and they were not on any teratogenic drugs.
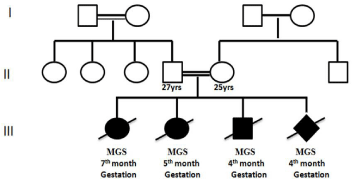
Figure 1: Pedigree of the family with Recurrent Meckel-Gruber syndrome
(MGS).
During her first pregnancy, ultrasound examination revealed occipital encephalocele, ventricular septal defect, polycystic kidneys and bilateral club foot features suggestive of Meckel-Gruber Syndrome (MGS). The couple were counselled about the condition and they opted for termination of the pregnancy at seventh month. Subsequently, in the second pregnancy antenatal ultrasound scan at fifth month illustrated occipital encephalocele, congenital heart defect, bilateral club foot and polycystic kidney. These features were suggestive of MGS and the couple opted for termination after non directive genetic counselling. Furthermore, third pregnancy ultrasound scan at fourth month revealed abnormal nuchal translucency thickness measuring 6.5mm, cerebral ventriculomegaly, bulky dysplastic kidneys and occipital skull defect indicative of MGS and the couple opted for termination.
Also in her present fourth pregnancy at 14 weeks of gestation, 3D and 4D ultrasound scan revealed echogenic kidneys, occipital encephalocele, club foot with polydactyly and also polydactyly of the hands, median cleft lip and palate in the foetus suggestive of MGS (Figure 2). The patient was counselled regarding the lethal outcome of MGS. The couple opted for termination of pregnancy and desired comprehensive genetic evaluation. Because of the occurrence of MGS in all the 4 pregnancies, we performed clinical exome sequencing of the fourth affected foetus using amniotic fluid. Since the first three fetuses with MGS were terminated, we could not perform molecular testing for them. Further, we performed parental karyotyping and TORCH profile (Toxoplasmosis, Rubella, Cyto Megalo Virus and Herpes Simplex Virus) in the mother.
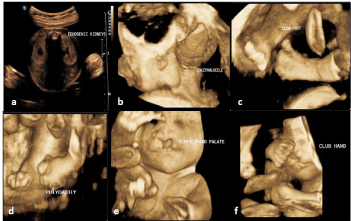
Figure 2: Ultrasound images of the fetus with MGS at 14 weeks of gestation
showing (a) Echogenic Kidneys (b) Encephalocele (c) Club Foot (d)
Polydactly (e) Cleft Lip and Palate (f) Club Hand.
Chromosomal analysis performed for both the parents was found to be normal. TORCH profile of the mother showed elevated IgG antibodies for Rubella. DNA sample from the amniotic fluid of the foetus was subjected to clinical exome sequencing using Illumina NGS platform for 10 genes (B9D1, B9D2, CC2D2A, CEP290, MKS1, NPH3, RPGRIP1L, TCTN2, TMEM216, and TMEM67) which were found to be associated with MGS. In addition, pathogenic and likely pathogenic variants in all other well covered genes present in the strand clinical exome panel were investigated for secondary and incidental findings. We found a novel heterozygous missense substitution (c.1949C>T, p.Pro650Leu) in exon17 of the TCTN2 gene in the foetus which alters a conserved residue in the protein. However, the clinical significance of this variant is unknown. Further in silico missense prediction tools (SIFT, LRT, Mutation Taster, Polyphen-2 and FATHMM) predicted that this variant is potentially damaging the protein function. Furthermore, clinical exome sequencing of other genes including pathogenic and likely pathogenic did not reveal any synonymous or non-synonymous variants. The study was approved by the Institutional ethics committee, Osmania University and the couple were investigated with informed written consent.
Discussion
Meckel Gruber Syndrome (MGS) is a lethal, autosomal recessive, multi systemic disorder, associated with mutations affecting ciliogenesis. MGS is typically a perinatal lethal syndrome characterised by posterior fossa abnormalities (most frequently occipital encephalocele), bilateral enlarged cystic kidneys, postaxial polydactyly and hepatic ductal plate malformation. Additional malformations such as microphthalmia, facial clefting, heart defects, fibrotic changes in the portal area of the liver, incomplete or ambiguous development of external or internal genitalia are often present [1]. MGS affects both genders equally and consanguinity has been reported to be an important factor in the genetic basis of the disease. Once diagnosed, the chances of MGS in subsequent pregnancy are 1 in 4 (25%) [2]. The worldwide incidence of MGS varies from 1 in 13,250 to 1, 40,000 live births [3].
The ciliopathies are a group of related inherited diseases characterized by malformations in multiple organ systems, with kidney, skeleton, and brain malformations frequently observed. MKS1, the first gene found to be mutated in MGS, encodes a ciliary protein. Subsequently, nine additional genes have been identified, all similarly encoding ciliary proteins (B9D1, B9D2, CC2D2A, CEP290, NPHP3, RPGRIP1L, TCTN2, TMEM216 and TMEM67) [4].
In the present study, we report a consanguineous couple giving birth to repeated MGS children. Clinical exome sequencing results identified c.1949C>T (p.Pro650Leu), a novel heterozygous missense variant in the TCTN2 in the foetus with MGS. TCTN2 is located on chromosome 12 region and encodes a type I membrane protein that belongs to the tectonic family, which might be involved in hedgehog signalling, and essential for ciliogenesis. A homozygous splice site mutation in TCTN2 was identified [5] in a consanguineous Saudi family with 5 pregnancies affected with MKS. Majority of the pathogenic variations in the TCTN2 gene associated with MGS are truncating variations. Furthermore, since we found no mutations in the 10 genes except for TCTN2, the present mutation could cause the symptoms of our patient suggesting that this mutation is a potentially important variant. The TCTN2 c.1949C>T (p.Pro650Leu) variant is extremely rare and was not reported in previous literature. The identified variant was found to alter a conserved residue and is predicted to be damaging the protein function essential for ciliogenesis. Since MGS is an autosomal recessive condition, the novel heterozygous missense variant identified in the TCTN2 gene might have increased the risk of MGS which could be due to dominantnegative effect.
In the present study, the couple had first degree consanguinity. In our previous study on risk factors for congenital anomalies in 3,301 High Risk Pregnant (HRP) women, consanguinity played a major role in HRP women with bad obstetric history [6]. In addition, there is evidence that the prevalence of MGS is higher in populations with higher consanguinity rates, particularly in India, Pakistan, Kuwait and other Arab countries [1].
Furthermore, TORCH analysis performed in the mother revealed raised IgG antibodies for Rubella. We have previously carried out a study on influence of TORCH infections in 1,158 HRP women and found that rubella infection predisposed HRP women to have foetal congenital malformation [7].
Conclusion
We report the presence of a novel heterozygous missense variant, c.1949C>T (p.Pro650Leu) in the TCTN2 gene using clinical exome sequencing in a family with recurrent MGS. We suggest that families with repeated ciliopathy phenotypes such as MGS, mutational analysis using a targeted clinical exome panel allows a rapid molecular diagnosis and provides important information for counselling the parents about recurrence risk, prognosis of the foetus and the outcome in the subsequent pregnancies.
References
- Barisic I, Boban L, Loane M, Garne E, Wellesley D, Calzolari E, et al. Meckel-Gruber Syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. European Journal of Human Genetics. 2015; 23: 746-752.
- Young ID, Rickett AB, Clerke M. High incidence of Meckel’s syndrome in Gujarati Indians. J Med Genet. 1985; 22: 301-304.
- Panduranga C, Kangle R, Badami R, Patil PV. Meckel-Gruber syndrome: Report of two cases. Journal of neurosciences in rural practice. 2012; 3: 56- 59.
- Shaheen R, Faqeih E, Alshammari MJ, Swaid A, Al-Gazali L, Mardawi E, et al. Genomic analysis of Meckel-Gruber syndrome in Arabs reveals marked genetic heterogeneity and novel candidate genes. European Journal of Human Genetics. 2013; 21: 762-768.
- Shaheen R, Faqeih E, Seidahmed MZ, Sunker A, Alali FE, AlQahtani K, et al. A TCTN2 mutation defines a novel Meckel Gruber syndrome locus. Hum Mutat. 2011; 32: 573-578.
- Sunitha T, Prasoona KR, Kumari TM, Srinadh B, Deepika ML, Aruna R, et al. Risk factors for congenital anomalies in high risk pregnant women: A large study from South India. Egyptian Journal of Medical Human Genetics. 2016.
- Prasoona K Rebekah, Srinadh B, Sunitha T, Sujatha M, Deepika MLN, Vijaya Lakshmi B, et al. Seroprevalence and Influence of Torch Infections in High Risk Pregnant Women: A Large Study from South India. The Journal of Obstetrics and Gynecology of India. 2015; 65: 301-309.