
Case Report
Ann Hematol Oncol. 2014;1(2): 1008.
Surgical Castration for the Late Development of Resistance to Medical Castration with LHRH Agonists and Antagonists in Castrate-Resistant Prostate Cancert
Yaser Al-Marrawi1*,Nazia Raja-Khan2, Kevin WuCarol Mallon RN OCN1, Monika Joshi1, Sheldon Holder1Matthew Kaag3,Jeffrey Sivik DPharm3 and Joseph J Drabick1
1Penn State University/Hershey Medical Center, Cancer Institute, Hershey, PA, USA
2Penn State University/Hershey Medical Center, Endocrinology Division, Hershey, PA, USA
3Penn State University/Hershey Medical Center, Urology Division, Hershey, PA, USA
4Penn State University/Hershey Medical Center, Pharmacy, Hershey, PA, USA
*Corresponding author: Yaser Al-Marrawi, Penn State University/Hershey Medical Center, Cancer Institute, Hershey, PA, USA, 500University Drive, Hershey, PA 17033
Received: October 10, 2014; Accepted: October 31, 2014; Published: November 03, 2014
Abstract
Acquired resistance to GNRH agonists or antagonists used for the induction of medical castration for the treatment of metastatic prostate cancer has been rarely reported. To date, the mechanism(s) behind the development of apparent resistance to these agents has yet to be adequately defined while the impact of this refractoriness may possibly be detrimental even in Castrate-Resistant Prostate Cancer (CRPC). Here we describe two men with CRPC who were compliant with their GNRH agonist and who had documented low testosterone levels for years. They then developed rising testosterone levels despite documented continued receipt of these agents. Switching to the pure LHRH antagonist, degarelix, did not improve the testosterone suppression. Of interest, repeated determinations of luteinizing hormone were undetectable in both men suggesting the apparent resistance was not at the level of the hypothalamicpituitary axis. They did not have elevated Beta-HCG, prolactin or estrogen levels. The testosterone levels also did not respond to abiraterone given for their CRPC nor did them everhave sterile abscesses which have been associated with resistance to LHRH agonists. They therefore underwent surgical castration with bilateral orchiectomy and their testosterone levels returned to appropriate castrate levels againsuggesting that the failure of medical castration was not due to androgen production by the tumor cells but rather by the gonads. Understanding the endocrine mechanism for this failure remains unclear but the response to surgical castration suggests it is at the level of the gonads. These cases and others support the continued checking of testosterone levels while on long term medical castration agent since resistance can occur after many years of good control. Then, surgical castration becomes the treatment of choice.
Keywords: Surgical Castration; LHRH Agonist Resistance; LHRH Antagonist Resistance; Medical Castration Resistance; Resistant Prostate Cancer; Endocrine Therapy Resistance; Prostate Cancer Resistant To Endocrine Therapy
Introduction
Androgen-Deprivation Therapy (ADT) in the form of castration has been the mainstay of therapy for prostate cancer since the 1940s [1-5]. The goal of ADT is to reduce gonadal androgens to castration levelswhether applied surgically with bilateral orchiectomy or medically via gonadotropin receptor- blockage using either LHRH agonists or antagonist (Figure 1). ADT typically only works for a limited time before prostate cancer cells find a way of surviving despite castration serum levels of testosterone, the prostate cancer cells may survive on low levels of androgens produced by the adrenal glands or may actually produce their own androgens [6-8]. They may also survive by becoming completely independent of androgen stimulation [9-11]. Prostate cancer at that juncture has become castration resistant. Even in CRPC, castration therapy should be continued since many of the clones remain sensitive to castration such that stopping it and allowing testosterone levels to rise can result in flares of disease [12]. It has been demonstrated that incomplete testosterone suppression has been associated with increased biochemical failure rates and metastases with worsesurvival [13]. Previous case reports reported a correlation between LHRH analogues resistance and formation of sterile abscesses or inflammation at sites of LHRH analogue injection which apparently interfere with absorption of the drug from the injection site [14]. However, other reports noted LHRH resistance despite absence of any sterile abscess with goserelin [15], and that resistance developed shortly after initiation of therapy with the second 3-month injection despite the sharp initial response in terms of serum testosterone level and PSA level as well. Many oncologists stop monitoring testosterone levels after levels of testosterone post initiation of medical castration have attained castrate levels. The previous case-reports and our experience suggest that testosterone levels should be monitored regularlyin patients on medical castration with GNRH agonists or antagonists since resistance to these drugs can occur either early or late. The mechanism for this apparent failure of these agents to adequately suppress testosterone after years of control has not been elucidated adequately to date but does respond to surgical castration. Here we present 2 cases of men with advanced prostate cancer who developed rising testosterone levels despite regular receipt of theseagents which had worked previously. Some endocrine testingwere performed trying to uncovera defectresponsible for this breakthrough and in both cases the problem resolved with surgical castration.
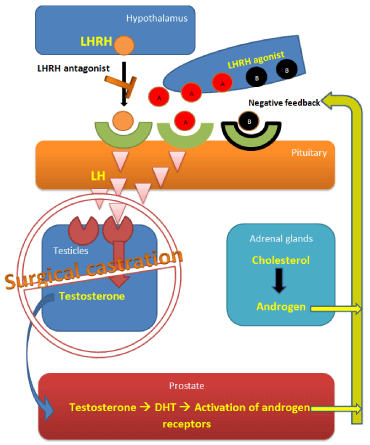
Figure 1: Physiology of the hypothalamus pituitary testes axis.
Case Reports
Case 1
The first patient is an 85year old gentleman who was originally diagnosed with BPH in 1994 with an elevated PSA. He underwent trans-urethral resection of the prostate and pathology was negative for malignancy. He started having a rapidly rising PSA late in 2004 up to 35. His bone scan on Nov. 5th, 2004 showed one solitary area of uptake in 10th left rib worrisome for metastatic focus. It was consistent with a sclerotic area on a plain film. Biopsy confirmed adenocarcinoma of the prostate. He was placed on Leuprolide depot at diagnosis in Nov. 2004 and his PSA declined and reached a nadir level of 2. His PSA and bone scan remained stable till May 2007 when the 10th riblesion appeared worse on bone scan but no additional lesions were noted; PSA at that time was slightly higher at 5.6 and he was asymptomatic.
He continued the leuprolide depot injections. He presented with gross hematuria in Nov. 2006 and underwent TURBT x 2 where he was found to havean incidental superficial bladder transitional cell carcinoma and he has been without any evidence of TCC recurrence ever since on regular surveillance.
Two months later in Jul. 2007, his bone scan revealed new rib metastatic foci and his PSA increased to 7.8. (See Table 1) His DEXA scan was consistent with osteoporosis. He was then diagnosed as having CRPC and so was started on high dose Bicalutamide as first line treatment in addition to his LHRH agonist and he was also started on monthly Zolidronic acid for the metastatic bone disease. On therapy, his PSA declined to 3.4. Testosterone was documented then to be in a castrate range at< 15 ng/dL on Apr. 1st, 2008 and a CT urogram revealed a large lobular prostate. PSA then started to rise slowly again. Bicalutamide was therefore stopped trying to induce an anti androgen withdrawal response; PSA, however only dropped from 5.52 to 5.35 with Bicalutamidewithdrawal and it began to raise again consistent with disease progression. Bone scan and CT scansin Jul. 2008 were done in lieu of rising PSA and they were both stable. He was then started on Ketoconazole and hydrocortisone as the next second line hormonal therapy. The PSA dropped on ketoconazolefrom 7.06 to 1.66 over 9 months and remained low. Zolidronic acid was stopped after 2 years of monthly treatments. He had another TURP meanwhile for large volume prostate with symptoms of outflow obstruction. Of interest, the prostate tissue obtained from this TURP was negative for cancerconsistent with an excellent response to ketoconazole. After this good control, his PSA started to rise slowly again, and therefore Ketoconazole was discontinued. Denosumab 120 monthly with Ca and vitamin D were started. Sipuleucel-T was offered to him but he declinedand therefore abiraterone was started with prednisone late in Apr. 2013 which he tolerated very well. His PSA dropped from 6.35 in Apr. 2013 to 3.58 in May 2013. However, his testosterone level was noted to be 80 on May 30th, 2013 and was 50 on Jun. 6th, 2013. It had been very low for years as noted. Thus, he was switched from Lupron to degarelix “Firmagon” in Jun. 2013. However, no improvement in testosterone level was noted as his testosterone level remained between 50-105 between Jul. 2013 and Feb. 2014 (Table 1),and therefore surgical bilateral orchiectomy was recommended. In an effort to understand the mechanism behind this surprising aberration we did some laboratory investigations. He never had any problems with his injections and there were no clinical or radiographic findings to suggest the development of sterile abscesses at the injection sites. His LH was low < 0.22mIU/ml in Dec. 2013 and also in Feb. 2014 (normal range is 1.14-8.75mIU/ml). His prolactin, estrogen and HCG serum levels were normal. In the interim, he had moreurinary output obstruction and a temporary urinary catheter was placed and was treated and resolved with flomax. On Mar. 10th, 2014, he underwent rigid cystoscopy with another transurethral resection of the prostate and also had bilateral scrotal orchiectomy under spinal anesthesia. His surgical pathology revealed high grade prostatic adenocarcinoma in bladder neck whereas his testicles showed global atrophy. On his first post-op visit on Apr. 24th, 2014, his PSA was stable on abiraterone at 4.82. His testosteronedid come down to < 15 by Apr. 2014; free testosterone was <1.He has continued to be stable clinically and byJun. 2014 his PSA dropped again on continued abiraterone to 3.88 suggesting the surgical castration may have led to better PSA control.
![]()
Date
PSA
Serum testosterone
BMI
Jan. 18th, 2012
3.09
< 15
28.36
Apr. 18th, 2012
3.68
10
28.46
Jun. 14th, 2012
2.42
< 5
28.46
Aug. 2nd, 2012
3.22
< 5
28.4
Oct. 5th, 2012
4.33
< 5
28.4
Nov. 30th, 2012
4.91
7
28.4
Jan. 7th, 2013
5.83
< 5
28.4
Feb. 7th, 2013
5.51
< 5
28.8
Apr. 2nd, 2013
6.35
< 5
28.98
May 31st, 2013
3.58
80
27.75
Jun. 6th, 2013
3.58
50à so luprolide was changed to degarelix
27.75
Jul. 7th, 2013
3.2
105
27.99
Oct. 4th, 2013
2.89
70
27.56
Dec. 6th, 2013
2.95
63
27.56
Feb. 4th, 2014
4.78
50
27.44
surgical castration Mar. 10th, 2014 with bilateral orchiectomy
27.28
Apr. 24th, 2014
4.82
< 5
27.44
June 20th, 2014
3.88
< 5
27.44
Table 1: PSA and Testosterone levels for Patient of Case 1.
Case 2
This is an 83 year old gentleman with history of spine degenerative joint and disk disease who had laminectomy complicated with osteomyelitis in 2010. On routine follow up imaging of the spine in Sep. 2011, he was noted to have multiple sclerotic lesions involving the spine worrisome for metastatic disease.He underwent biopsies of some of the lytic lesions on Oct. 17th, 2011. The pathology revealed an adenocarcinoma consistent with prostatic carcinoma by immunohistochemistry. Bone scan showed additional multiple osseous lesions involving ribs and spine. His initial PSA at that time was elevated at 16.5. His last PSA, checked several years prior, was about 4 which had been stable compared with prior years. Therefore, he was seen by urology and medical oncology and was started onLeuprolide depot injection and bicalutamide as total androgen blockade in Oct. 2011. He also began denosumab for the osseous metastatic disease in Feb. 2012. His PSA was suppressed until Jul. 2012 when it rose to 16.84while still in castrate state by measuring testosterone level consistentwith the development of CRPC. The patient had multiple significant medical problems including cardiomyopathy with intermittent bouts of CHF. Bicalutamide was stopped on Jul. 10th, 2012 and we hoped for an anti-androgen withdrawal response. Unfortunately, his PSA continued to climb and he thus did not respond to bicalutamide withdrawal (see Table 2). Sipuleucel-T could not be administered due to cardiac comorbidities particularly with leukapheresis requirement. He was then started on abiraterone with low dose prednisone on Aug. 16th, 2012. He underwent CT scans on Feb. 25th, 2013 which showed new bladder lesions and disease progression in bones. He was then re-evaluated by urology with cystoscopy and was found to have new bladder cancer. He underwent Transurethral Resection of the Bladder Tumor (TURBT) on Mar. 26th, 2013. Surgical pathology revealed high grade papillary Transitional Cell Carcinoma (TCC), Ta G4 and was treated with BCG treatments. His PSA had been slowly rising between 16 and 32 till May 2013 when his PSA rose from 32.57 to 46.48. His testosterone level was noted to be 100 despite having had Leuprolide (the 3 month depot) 2 months prior and despite being on abiraterone and prednisone as well. Previous testosterone levels while on abiraterone were running about 48, much higher than expected with Leuprolide and abiraterone use. Therefore, on May 17th, 2013 Leuprolide was changed to degarelix in an effort totry to obtain adequate testosterone suppression, while abiraterone was continued. His testosterone level continued to be high on degarelix as well as it rose to 123 (Table 2). Therefore, it too was stopped on Sep. 19th, 2013 and surgical castration with bilateral orchiectomy was recommended. LH levels were low and no other endocrine abnormalities were found. This surgical castration was done along with his surveillance cystoscopy on Nov. 1st, 2013. The testicles showed simple atrophy. His post-op testosterone level came down to < 5 and have remained appropriately low, however, his PSA continued to rise as it rose from 88.7 in Nov. 2013 to 97.1 in Jan. 2014 to 142.16 in Mar. 2014 consistent with continued disease progressionon abiraterone. Therefore his abiraterone was stopped and was started on Enzalutamide on Dec. 15th, 2013. His CT scans and bone scan in Jan. 2014 were consistent with stable disease. Thealkaline phosphatase started to rise suggestive of worsening bone disease. His PSA rose further to 151.9 in May 2014and therefore repeated CT scans were done which revealed stable osseous metastatic disease and patient elected to start Radium 223 on May 8th, 2014; he was having some increasing bone discomfort at the time. He received 3 Radium doses so far to the date of writing this publication but his PSA continued to rise despite being constantly in the surgical castration state with testosterone level &5 since the bilateral orchiectomy. He remains pain free however at the current time. The plan is to offer Docetaxel post Radium for disease progression.
![]()
Date
PSA
Serum testosterone
BMI
Mar. 8th, 2012
13.25
16
25.45
Jul. 9th, 2012
16.84
16
25.17
Aug. 16th, 2012
22.4
63
25.04
Sep. 14th, 2012
22.8
70
25.71
Nov. 6th, 2012
21.69
31
25.71
Dec. 12th, 2012
25.59
48
25.49
Feb. 26th, 2013
32.57
48
25.64
May 1st, 2013
46.48
100
24.98
May 17th, 2013
36.35
78à so luprolide was changed to degarelix
24.98
Jul. 25th, 2013
57.61
123
25.68
Sep. 15h, 2013
59.86
Not done
25.26
Nov. 12th, 2013
88.70
48
25.97
surgical castration Nov. 1st, 2013 with bilateral orchiectomy
25.97
Jan. 14th, 2014
97.12
<5
26.13
Mar. 31st, 2014
142.16
12
26.13
May 5th, 2014
151.49
5
24.65
Jun. 2nd, 2014
176.46
<5
24.4
Jul. 1st, 2014
264.26
Not done
24.01
Table 2: PSA and Testosterone levels for Patient of Case 2.
Discussion
We described two men who had initial long term excellent testosterone suppression on LHRH agonists but later developed rising then persistently higher non-castrate testosterone levels despite continued receipt of these agents. We tried to explore the mechanisms for this late failure. Both men had repeatedly low LH levels suggesting the agents were indeed working at the pituitary to suppress production. There were no sterile abscesses noted which have been reported to impair absorption of the drugs at the injections sites and the low LH levels suggested that the drug was indeed absorbed and working at least at the level of the pituitary so mechanisms such as alternation in the GNRH receptor or antibodies interfering with GNRH drugs should not be operant. Likewise, there were no other abnormalities in estrogen, BHCG or prolactin noted. Changing from the GNRH agonist, leuprolide to the pure GNRH antagonist, degarelix, had no impact at all. Surgical castration, however, did result in return of testosterone levels to castrate levels. This suggested to us that the source of resistance to these drugs was at the level of the gonad. The removed testicles just showed atrophy on pathologic analysis. We postulated that in a state of chronic low LH levels the gonads may become independent of LH completely, or the receptors for LHin the gonads may become hypersensitive to even small amounts of LH; and this, ineither case, results in increased testosterone production. Another possibility to consider would be the presence of LH-like peptide or even beta-HCG driving the gonadal production of testosterone. Beta-HCG is known to act on the LH receptors in the testicles and therefore can stimulate testosterone secretion. It is possible that some prostate cancer cells may start secreting beta HCG (or some other LH-like substances) in a paraneoplastic fashion that can stimulate LH receptors in testes. Our patients had normal beta HCG determinations but we cannot rule out the presence of LH or HCG like peptides which are functional but not measured. Surgical castration would also work if this was the mechanism as well. We also considered that the results could have been due to a laboratory artifact due to the cross reactivity of the testosterone assay with other androgen-like molecules. We wondered if the abiraterone, which has steroidal hormonal structure would do that. We thought this unlikely, however since we have many men on LHRH drugs and abiraterone in whom we regularly monitor testosterone levels and have only observed this phenomenon rarely and also because the tester one levels dropped post castration but while still on abiraterone. Likewise, in clinical studies with abiraterone, testosterone levels were documented to be very low consistent with its mechanism of action [16,17]. Whether the transient non-castrate levels of testosterone noted in our patients had an additional adverse effect in the course of their already CRPC is debatable but Patient 1 did have a lower PSA coincident with a lower testosterone after surgical castration.
Synthetic LHRH analogues have been used to provide medical castration in prostate cancer with progression after surgical prostatectomy or prostatic radiotherapy and with metastatic disease at diagnosis as in the men described here. Historically, the goal of medical castration is to achieve serum testosterone levels less was than 50 ng/dl (<1.7nmol/L) [18]. However, Testosterone levels >32 ng/dL (>1.1 nmol/L) are currently considered no longer fully suppressed according to a recent report [19]. With surgical orchiectomy, testosterone levels should reach <20 ng/dL (< 0.7 nmol/L) [20]. LHRH agonists and antagonists has been an excellent non-invasive therapeutic option for castration and appear equally effective in comparison to surgical orchiectomy in clinical outcomes. However, in about 13%-21% of patients, testosterone levels with LHRH agonist may not achieve the surgical orchiectomy testosterone levels and indeed higher testosterone levels have been associated with increased levels of PSA and an increased death rate in patients with CRPC [13,20]. On the other hand, only 10% of patients who undergo surgical castration will fail to achieve total testosterone level less than 20 ng/dL (<0.7 nmol/L) [21]. Previous studies have shown that up to 12% of prostate cancers patients fail to have testosterone level to less than 50 ng/dl (<1.7 nmol/L) [22-24]. A report of 451 patients showed that percentage was 11% [25]. In 2010, the largest study to date, presented a population of 2196 patients which confirmed that breakthrough can occur with lower degrees of testosterone suppression per patient of 3.4% = 50 ng/dL (=1.7 nmol/L) and 6.6% = 32 ng/dL (=1.1 nmol/L) using testosterone assays that have been shown to be more accurate at low levels [26]. By its effect on both adrenal and tumoral hormonogenesis, abiraterone allows clinicians to bring serum testosterone levels to less than 20 ng/dl and in theory help some of those patients who may be incompletely suppressed by LHRH analogues. Of note, it did not do so in our patients on the drug suggesting they still had gonadal production of male hormones not suppressed by abiraterone. LH expression in prostate cancer tissues has been associated with poor prognosis, while FSH has been shown to stimulate prostate cell growth in hormone-refractory prostate cancer cell lines. In addition, there is evidence that high testosterone levels are associated with poor prognosis [19,27]. Patients with a higher than castration levels of testosterone at 6 months experienced a 1.3-fold greater risk of cancer-related mortality (95%CI 1.05-1.69; p< 0.05) [13]. GnRH and its receptors are present in multiple other tissues in addition to the pituitary gland. They are found in prostate, ovaries, breasts, placenta and endometrium. Testosterone breakthrough risk was studied in 2196 patients from the British Columbia Cancer registry data set which revealed the risk of testosterone breakthrough to >1.1 nmol/L (>32 ng/dL) is 6.6%, whereas is it 5.4 % for level > 50 ng/dL (>1.7 nmol/L) per patient course. On the other hand, that risk is 5.4% and 2.2% respectively per LHRH agonist injection. Recurrence of the breakthrough decreases the Prostate cancer-free survival from 73% to 58% [26]. Expression of LH and Beta-HCG in prostate cancer tissues is consistent with poor prognosis and entitles higher risk for metastatic disease. LH upregulates enzymes and genes responsible for sex steroids production via prostate cancer cells [28]. This can lead to an important role of LH-receptor signaling in prostate cancer in becoming castration resistant. However, LH levels were not noted to be higher in prostate cancer when compared with those levels in benign prostatic hypertrophy or even when those levels are compared between localized and locally advanced prostate cancers [29-31]. Furthermore, this expression of either LH or beta-HCG could be responsible for more aggressive malignant evolution of prostate cancer cells by changing their shapes, migration, invasiveness and adhesiveness as well [32]. Differently, FSH was noted in previous research to be higher in prostate cancer than in BPH. It was also found to be even higher in locally advanced prostate cancer than in localized prostate cancer [29,33]. FSH stimulates prostate cell growth in castration resistant prostate cancer cell lines [34]. In absence of prostate cancer, FSH synthesis and release can be suppressed by a protein produced by prostate gland called PIP (Prostatic In hibin Peptide). This PIP production is suppressed in prostate cancer. Low PIP allows more FSH to be synthesized and released and in turn more prostate cancer cell growth [35-37]. Our patients had low LH levels suggesting that the GNRH agonists and antagonists used were effective in suppressing this intermediary hormone but not the testosterone production at the level of the gonads. No published data explaining the reason for treatment failure with LHRH agonists or antagonists exist to the best of our knowledge. Similarly, no identified risk factors for developing LHRH therapy resistance have been described. Our experience suggests that failure of one LHRH agonist or antagonist to suppress testosterone to castration level reflects a general resistance to LHRH therapy rather than only resistance to the agent in question. Many oncologists stop monitoring testosterone level after reaching first castrated level of testosterone post initiation of medical castration. The two cases we presented here stress the necessity of monitoring testosterone level periodically while on endocrine therapy but particularly with disease progression. Patients who develop sterile abscesses or inflammation at sites of depot injectionsshould have close monitoring of their testosterone level. Resistance to GNRH agents appears to be at the level of the gonads and bilateral surgical orchiectomysucceeds in getting very low testosterone levelsin men who have failed all different forms of castration from LHRH agonist, LHRH antagonist, androgen receptor inhibitors, and CYP 17 hydroxylase inhibitors. Evidence suggests that this may benefit patients even in the castrate-resistance phase of the disease therapy.
References
- Perlmutter MA, Lepor H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev Urol. 2007; 9: S3-8..
- Janknegt RA, Abbou CC, Bartoletti R, Bernstein-Hahn L, Bracken B, Brisset JM, et al. Orchiectomy and nilutamide or placebo as treatment of metastatic prostatic cancer in a multinational double-blind randomized trial. J Urol. 1993; 149: 77-82..
- 3. Trachtenberg J. Hormonal management of stage D carcinoma of the prostate. Carson CC, editor. In: Problems in Urology. Philadelphia: JB Lippincott Co. 1993; 215–225..
- Ciezki JP, Klein EA, Angermeier K, Ulchaker J, Chehade N, Altman A, et al. A retrospective comparison of androgen deprivation (AD) vs. no AD among low-risk and intermediate-risk prostate cancer patients treated with brachytherapy, external beam radiotherapy, or radical prostatectomy. Int J RadiatOncol Biol Phys. 2004; 60: 1347–1350. .
- 5. Huggins C, Hodges CV. Studies on prostatic cancer II: the effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941; 43: 209–223. .
- Devlin HL, Mudryj M. Progression of prostate cancer: multiple pathways to androgen independence. Cancer Lett. 2009; 274: 177-186..
- Schröder FH. Progress in understanding androgen-independent prostate cancer (AIPC): A review of potential endocrine-mediated mechanisms. European Urology. 2008; 53: 1129-1137. .
- Dillard PR, Lin MF, Khan SA. Androgen-independent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol. 2008; 295: 115-120..
- Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997; 57: 314-319..
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995; 332: 1393-1398..
- Craft N, Chhor C, Tran C, Belldegrun A, DeKernion J, Witte ON, et al. Evidence for clonal outgrowth of androgen-independent prostate cancer cells from androgen-dependent tumors through a two-step process. Cancer research. 1999; 59: 5030-5036. .
- 12. Zelefsky MJ, Eastham JA, Sartor AO. Castration-Resistant Prostate Cancer. Vincent T. DeVita J, Lawrence TS, Rosenberg SA, editros. In: DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology, 9e. Philadelphia, PA: Lippincott Williams & Wilkins. 2011. .
- Perachino M, Cavalli V, Bravi F. Testosterone levels in patients with metastatic prostate cancer treated with luteinizing hormone-releasing hormone therapy: prognostic significance?. BJU Int. 2009; 105: 648-651. .
- Curry EA 3rd, Sweeney CJ. Resistance to luteinizing hormone releasing hormone agonist therapy for metastatic prostate cancer. J Urol. 2002; 168: 193..
- Daskivich TJ, Oh WK. Failure of gonadotropin-releasing hormone agonists with and without sterile abscess formation at depot sites: insight into mechanisms? Urology. 2006; 67: 1084..
- Ryan CJ, Molina A, Li J, Kheoh T, Small EJ, Haqq CM, et al. Serum androgens as prognostic biomarkers in castration-resistant prostate cancer: results from an analysis of a randomized phase III trial. J ClinOncol. 2013; 31:2791-2798. .
- Ryan CJ, Peng W, Kheoh T, Welkowsky E, Haqq CM, Chandler DW, et al. Androgen dynamics and serum PSA in patients treated with abiraterone acetate. Prostate Cancer Prostatic Dis. 2014; 17: 192-198..
- Oefelein MG, Resnick MI. Effective testosterone suppression for patients with prostate cancer: is there a best castration? Urology. 2003; 62: 207-213..
- Morote J, Orsola A, Planas J, Trilla E, Raventós CX, Cecchini L, et al. Redefining clinically significant castration levels in patients with prostate cancer receiving continuous androgen deprivation therapy. J Urol 2007; 178: 1290-1295. .
- Oefelein MG, Feng A, Scolieri MJ, Ricchiutti D, Resnick MI. Reassessment of the definition of castrate levels of testosterone: implications for clinical decision making. Urology. 2000; 56: 1021-1024..
- Gomella LG. Effective testosterone suppression for prostate cancer: is there a best castration therapy? Rev Urol. 2009; 11: 52-60..
- Morote J, Planas J, Salvador C, Raventós CX, Catalán R, Reventós J. Individual variations of serum testosterone in patients with prostate cancer receiving androgen deprivation therapy. BJU Int. 2009; 103: 332-335..
- 23. Thombal B, Berges R. Optimal control of testosterone: a clinical case-based approach of modern-androgen-deprivation therapy. EurUrolSuppl. 2008; 7: 15-21. .
- Morote J, Esquena S, Abascal JM, Trilla E, Cecchini L, Raventós CX, et al. Failure to maintain a suppressed level of serum testosterone during long-acting depot luteinizing hormone-releasing hormone against therapy in patients with advanced prostate cancer. UrolInt. 2006; 77: 135-138. .
- 25. Cabeza A, Gomez A, Vilar S et al. Assessing the incidence of escapes of testosterone in the treatment with LHRH analogs: escape study. ASTRO Annual Science Meeting; 2009; Chicago: International Journal of Radiation Oncology, Biology, Physics. 2009; S355. .
- Pickles T, Hamm J, Morris WJ, Schreiber WE, Tyldesley S. Incomplete testosterone suppression with luteinizing hormone-releasing hormone agonists: does it happen and does it matter? BJU Int. 2012; 110: E500-507..
- Shaneyfelt T, Husein R, Bubley G, Mantzoros CS. Hormonal predictors of prostate cancer: a meta-analysis. J Clin Oncol. 2000; 18: 847-853..
- Pinski J, Xiong S, Wang Q, Stanczyk F, Hawes D, Liu SV. Effect of luteinizing hormone on the steroidogenic pathway in prostate cancer. Prostate. 2011; 71: 892-898..
- Sofikerim M, Eskicorapci S, Oruç O, Ozen H. Hormonal predictors of prostate cancer. Urol Int. 2007; 79: 13-18..
- Hilz H, Graefen M, Noldus J, Hammerer P, Knabbe C, Huland E, et al. Advanced prostate cancer is associated with a decrease in serum luteinizing hormone. Eur Urol. 2000; 38: 243-249..
- Heracek J, Urban M, Sachova J, Kuncova J, Eis V, Mandys V, et al. The endocrine profiles in men with localized and locally advanced prostate cancer treated with radical prostatectomy. Neuro Endocrinol Lett. 2007; 28: 45-51..
- Wu W, Walker AM. Human chorionic gonadotropin beta (HCGbeta) down-regulates E-cadherin and promotes human prostate carcinoma cell migration and invasion. Cancer. 2006; 106: 68-78..
- Alam H, Maizels ET, Park Y, Ghaey S, Feiger ZJ, Chandel NS, et al. (2004) Follicle-stimulating hormone activation of hypoxiainducible factor-1 by the phosphatidylinositol 3-kinase/AKT/Ras homolog enriched in brain (Rheb)/mammalian target of rapamycin (mTOR) pathway is necessary for induction of select protein markers of follicular differentiation. J BiolChem. 2004; 279: 19431–19440..
- Ben-Josef E, Yang SY, Ji TH, Bidart JM, Garde SV, Chopra DP, et al. Hormone-refractory prostate cancer cells express functional follicle-stimulating hormone receptor (FSHR). J Urol. 1999; 161: 970-976..
- Garde S, Sheth A, Porter AT, Pienta KJ. Effect of prostatic inhibin peptide (PIP) on prostate cancer cell growth in vitro and in vivo. Prostate. 1993; 22: 225-233..
- Dowling CR, Risbridger GP. The role of inhibins and activins in prostate cancer pathogenesis. Endocr Relat Cancer. 2000; 7: 243-256..
- Bilezikjian LM, Vale WW. The Local Control of the Pituitary by Activin Signaling and Modulation. Open Neuroendocrinol J. 2011; 4: 90-101..