
Case Report
Ann Hematol Onco. 2023; 10(2): 1422.
CML with De Novo NPM1 Mutation and Rapid Progression to Myeloid Blast Cri-Sis despite Treatment with Imatinib: A Case Report
Chaudhry B¹; Parker J¹; Altohami M²; Borghi I²
1HDepartment of Haematology, Northampton General Hospital, UK
2Department of Haematology, Leicester Royal Infirmary, Infirmary Square, UK
3Imperial Molecular Diagnostics, Hammersmith Hospital, UK
*Corresponding author: Bilal Chaudhry Haematology Department, Northampton General Hospital, Northampton NN1, 5BD, UK. Email: bilal.chaudhry4@nhs.net
Received: April 15, 2023 Accepted: May 12, 2023 Published: May 19, 2023
Abstract
In this case we present a 41 year old male with chronic myeloid leukaemia who despite scoring low risk for Sokal progressed rapidly to blast crisis without warning and after having achieved a partial response to Imatinib therapy. We investigated using retrospective Next Generation Sequencing (NGS) and identified a novel NPM1 mutation at diagnosis and blast crisis together. We also found tyrosine kinase domain mutations using NGS not found with Sanger sequencing. We believe this case highlights the effectivity of NGS in identifying novel and existing mutations with higher sensitivity, which could help improve existing practice in prognostication and management of CML disease in patients not responding to treatment adequately.
Keywords: CML and NPM1; CML-BP; Next generation sequencing in CML; Imatinib failure; Blast crisis
Introduction
Chronic Myeloid Leukaemia (CML) is characterised by a triphasic natural history with Chronic (CP), Accelerated (AP) and Blast crisis (BP) phases. Contemporary therapeutic approaches based on Tyrosine Kinase Inhibitors (TKIs) have reduced the annual risk of progression to BP from 20% to around 1 to 1.5%, resulting in a vast improvement in quality of life and overall survival [1].
CML is characterised by the presence of a ‘Philadelphia chromosome’ due to a translocation between chromosome 9 and 22 t(9;22)(q34;q11). This results in production of the BCR/ABL fusion protein with resultant increased tyrosine kinase activity [2,3].
Current prognostic scoring systems include Sokal and more recently, the EUTOS Long-Term Survival score (ELTS). This predicts survival for patients on TKIs and may be more discriminatory than Sokal [4,5]. Although current prognostic scoring systems do not use cytogenetic information to predict outcome, the presence of additional Chromosomal Aabnormalities (ACA) such as trisomy 8 suggest an increased risk of progression to AP or BC [6].Despite the potential increased risk however, initial management remains the same [3,7].
Case Presentation
We report a 41 year-old man diagnosed with CML-CP, who was classed low risk for Sokal, EUTOS and ELTS at diagnosis. He achieved a partial response to imatinib therapy at three months on cytogenetics which was classed in ]accordance with European Leukaemia Net (ELN) 2013 guidance as optimal respon, se to first line TKI therapy, and did not indicate treatment failure [2]. Despite this he progressed to blast crisis one month later. Retrospective testing revealed mutated NPM1 at diagnosis and again at blast crisis.
In August 2019 a 41 year-old gentleman was referred urgently to haematology following an abnormal full blood count and blood film suggestive of CML. Other than a raised body mass index, examination was normal with no splenomegaly. There was no smoking or drinking history except for tobacco chewing and no relevant family history.
A bone marrow aspirate was consistent with CML-CP. Chromosomal analysis showed isolated 46,XY,t(9;22)(q34;q11) in 60% of cells and an additional chromosome 8 (trisomy 8) in 40% of cells. e13a3 BCR-ABL transcripts were detected by multiplex PCR and Q-PCR. Tyrosine kinase domain (TKD) mutation analysis was not carried out at diagnosis as per national practice [7]. He was deemed low risk according to Sokal (0.53), EUTOS (70.9), ELTS (0.817) and commenced on Imatinib 400mg daily rather than a 2nd generation TKI especially as his cardiac risk (QRisk3) was increased (relative risk 3.2) [9].
Molecular and cytogenetic response was assessed at 3 months as per standard national practice, following initiation of Imatinib therapy. Cytogenetics showed 3% of cells 46,XY,t(9;22)(q34;q11) and 97% demonstrating a normal karyotype. The trisomy 8 clone seen at diagnosis was not detected. BCR-ABL transcripts were significantly reduced (1498441 to 126331) and the BCR-ABL/ABL ratio was 43.413%.
Within days of receiving cytogenetic and molecular results, the patient re-presented with severe headaches, leukocytosis and blasts on a blood film. A bone marrow aspirate confirmed the presence of 25% myeloblasts and cytogenetics showed 47,XY,+8,t(9;22)(q34;q11) confirming progression to CML-BP (blast phase). The BCR-ABL/ABL ratio was 390.939%. Sanger sequencing performed across the Tyrosine Kinase Domain (TKD), detected a p.Gly250Glu (c.749G>A) G250E point mutation. There was no evidence of Central Nervous System (CNS) leukaemia on imaging or on Cerebrospinal Fluid (CSF) analysis.
He was treated with FLAG-IDA chemotherapy with additional CNS-directed therapy. Imatinib was changed to Nilotinib 400mg BD in view of the G250E point mutation and sensitivity to Nilotinib. A repeat bone marrow aspirate following blood count recovery showed 2% CD117+, CD34+ myeloid blasts. Cytogenetics showed normal karyotype consistent with complete cytogenetic remission and the BCR-ABL/ABL ratio had fallen to 2.4%.
He developed prolonged pancytopenia following a second cycle of FLAG-IDA plus Nilotinib therapy in February 2020. A repeat bone marrow sample post-chemotherapy showed 0.2% blasts on flow cytommetry, normal cytogenetics and a BCR-ABL/ABL ratio of 0.01%.
The COVID-19 pandemic in early 2020 caused significant delays with his planned allogeneic stem cell transplant. He required GCSF for ongoing significant neutropenia during the height of the first wave of the COVID-19 pandemic although his Measurable Residual Disease (MRD) during this time remained negative. After undergoing reduced-intensity conditioning allogeneic stem cell transplantation in August 2020 he remains alive and well in complete molecular remission, despite catching COVID-19 post-transplant.
This patient had progressed from CML-CP to CML-BP within 4 months following diagnosis despite scoring as low risk on current recommended prognostic scoring systems and after achieving a partial response on imatinib one month earlier without evidence of treatment failure. This seemed a very rapid progression of events, hence we retrospectively investigated his disease for additional genetic mutations using Next Generation Sequencing (NGS) for extended myeloid testing on all peripheral blood and bone marrow samples previously sent for BCR-ABL transcript monitoring.
Interestingly, mutated NPM1 was found in the absence of FLT3-ITD at CML diagnosis in July 2019. The peripheral blood sample three months after starting Imatinib was negative for mutated NPM1. At progression to CML-BP, one month later, mutated NPM1 was detected again and both G250e and T315I point mutations were found in the ABL1 gene, although only the G250e mutation was reported on conventional Sanger sequencing at the time of CML-BP progression.
Following his first cycle of FLAG-IDA chemotherapy in January 2020 his NPM1 status became negative again and no further TKD mutations were detected. Timeline of events can be seen in Table 1 with mutation percentage overtime in figure 1.
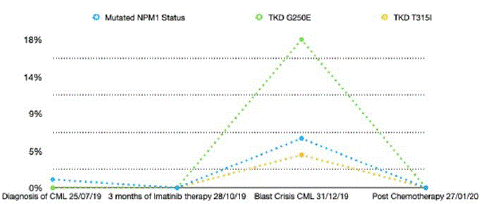
Figure 1: Variant Allele Frequency percentage (VAF) of gene mutations overtime for NPM1 and G250E, T315I in the TKD domain.
![]()
Timeline
Mutation status
Karyotype
BCR-ABL Transcripts
BCRABL/ABL %
July/August 2019 At CML diagnosis
NPM1mutFLTneg
46XY,t(9;22)(q34;q11) [60%], 47XY,t(9;22)(q34;q11) (+8) [40%]
1498441
361.07
October 2019 At 3 months Imatinib monitoring
NPM1negFLTneg
46XY,t(9;22)(q34;q11) [3%],
46XY [97%]126331
43.413
December 2019 at blast crisis (CML-BP)
NPM1mutFLTneg
G250E and T315I point mutations
47XY,t(9;22)(q34;q11) (+8) [95%]
46XY [5%]6841426
390.939
January 2020 after 1st cycle of Intensive Chemotherapy
NPM1negFLTneg
46XY [100%]
2151
2.436
June 2020 after 2nd cycle of intensive chemotherapy on nilotinib
NPM1negFLTneg
-
0
0
Table 1: Timeline of events for disease with NPM1 status, karyotype and BCR-ABL overtime. Cytogenetics were taken from bone marrow aspirate samples. BCR-ABL transcripts were tested from peripheral blood sampling taken at the same time as bone marrow samples.
Discussion
Presence of mutated NPM1 is a rare event with few cases reported to date in primary CML disease. No cases known to date have reported the presence of mutated NPM1 at CML-CP diagnosis and CML-BP together.
Out of the handful of cases known, Georgiou et al reported a CML patient with complete cytogenetic and molecular response to Imatinib who later developed NPM1 AML in a Ph-ve clone with BCR-ABL transcripts undetectable at relapse. In this case AML may have developed in events distinct from CML as de novo disease or possibly Imatinib therapy-induced AML [10]. A second case by Piccaluga et al reported cytoplasmic mutated NPM1 in a patient found at blast crisis only with no additional ACA’s. Authors thought this to be one of the drivers for AML transformation [11].
Known as an early founding event in AML pathogenesis, wild-type NPM1 functions as a shuttling protein between the nucleus and cytoplasm binding to other proteins and is thought to have a tumour suppressive function. Mutated NPM1 delocalises from the nucleus to the cytoplasm, resulting in down regulation of HOX genes and subsequent AML pathogenesis, although it’s exact role remains unclear [12].
Of the other adverse known risk factors, Trisomy 8 is classed as a ‘major route’ ACA by ELN and is known to confer adverse prognosis [3,13]. Trisomy 8 is likely to contribute towards CML progression with or without NPM1 mutation. Trisomy 8 in AML is thought to be associated with genetic changes which may have a stronger implication for leukemogenesis [14].
We postulate that mutated NPM1 at diagnosis in this patient likely conferred higher risk for blast transformation. In addition mutated NPM1 alongside ACA trisomy 8 and the BCR-ABL translocation may have played a synergistic role in accelerated disease. The recurrence of BCR-ABL, trisomy 8 and mutated NPM1 at CML-BP again suggest a possible link in a synergistic pathogenesis.
In terms of sensitivity with testing in CML disease, BCR-ABL1 mutations can be detected with sensitivities of around 20% with the conventional Sanger sequencing used in our patient. Greater sensitivity with NGS of about 3% permits early detection of clinically relevant BCR-ABL resistance mutations but not recommended in ELN guidance at diagnosis [3,15]. NGS performed at CML-BP in our patient revealed a T315I mutation not detectable using conventional techniques at time of blast crisis, highlighting importance of performing NGS in patients not responding adequately to TKI.
We also postulate that there is a chance more than one clone was present at diagnosis, with the Imatinib sensitive clone having been treated successfully by TKI. An additional NPM1 positive BCR-ABL, trisomy 8 clone may have undergone clonal evolution, contributing to the rapid sequence of events in disease progression. We also understand if there was more than a single clone, progression may have developed from TKI resistance to G250e and T315I point mutations either within the NPM1 mutated clone, +8 clone or in unrelated clones.
Current prognostic scoring systems do not account for clonal instability caused by genetic aberrations or mutations. Our patient’s diagnosis also predates the 2020 ELN guidance in which any ACA was deemed to be managed as ‘high risk’. ELN now classify ACA’s as ‘high-risk’ with poorer response to TKIs, however upfront management remains the same although more frequent molecular, cytogenetic and tyrosine kinase domain analysis testing is recommended [3]. We believe that further risk profiling ‘high risk’ patients at diagnosis could be achieved with greater sensitivity of Next Generation Sequencing (NGS) upfront. This could be used to detect myeloid gene mutations and TKD mutations at baseline and in patients not adequately responding to TKI therapy which would help guide prognostication, initial drug choice and improve CML disease monitoring.
References
- Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127: 2391-2405.
- Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, et al. European Leukemia Net recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013; 122: 872-884.
- Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020; 34: 966-984.
- Hu B, Savani BN. Impact of risk score calculations in choosing front-line tyrosine kinase inhibitors for patients with newly diagnosed chronic myeloid leukemia in the chronic phase. Eur J Haematology. 2014; 93: 179-186.
- Pfirrmann M, Baccarani M, Saussele S, Guilhot J, Cervantes F, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia. 2016; 30: 48–56.
- Fabarius A, Kalmanti L, Dietz CT, Lauseker M, Rinaldetti S, et al. Impact of unbalanced minor route versus major route karyotypes at diagnosis on prognosis of CML. Ann Hematol. 2015; 94: 2015–24.
- Smith G, Apperley J, Milojkovic D, Cross NCP, Foroni L, et al. A British Society for Haematology Guideline on the diagnosis and management of chronic myeloid leukaemia. Br J Haematol. 2020; 191: 171-193.
- National Institute for Health and Care Excellence. Guidance on the use of Imatinib for Chronic Myeloid Leukaemia [TA70]. 2013.
- Hippisley-Cox J, Coupland C, Brindle P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: prospective cohort study BMJ. 2017; 357: j2099.
- Georgiou G, Efthymiou A, Vardounioti I, Boutsikas G, Angelopoulou MK, et al. Development of acute myeloid leukemia with NPM1 mutation, in Ph-negative clone, during treatment of CML with imatinib. Leukemia. 2012; 26: 824–826.
- Piccaluga PP, Sabattini E, Bacci F, Agostinelli C, Righi S, et al. Cytoplasmic mutated nucleophosmin (NPM1) in blast crisis of chronic myeloid leukaemia. Leukemia. 2009; 23: 1370–1371.
- Brunetti L, Gundry MC, Sorcini D, Guzman AG, Huang YH, et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer cell. 2018; 34: 499-512.e9.
- Wang W, Cortes JE, Lin P, Khoury JD, Ai D, et al. Impact of trisomy 8 on treatment response and survival of patients with chronic myelogenous leukemia in the era of tyrosine kinase inhibitors. Leukemia. 2015; 29: 2263-6.
- Hemsing AL, Hovland R, Tsykunova G, Reikvam H. Trisomy 8 in acute myeloid leukemia. Expert Rev Hematol. 2019; 12: 947-958.
- Soverini S, Bavaro L, De Benedittis C, Martelli M, Iurlo A, et al. Prospective assessment of NGS-detectable mutations in CML patients with non-optimal response: the NEXT-in-CML study. Blood. 2020; 135: 534–541.