
Mini Review
Ann Hematol Onco. 2024; 11(1): 1446.
Intravascular Large B-Cell Lymphoma: A Report of Two Cases and Literature Review. What’s Next?
Meléndez MR¹; Cadavid N¹; Véliz AS¹; Merino B²; López-Rodríguez M²; Hidalgo F²; Perna C¹; Pian H¹; Moreno E¹; García-Cosío M¹*
1Department of Pathology, Ramón y Cajal University Hospital, Spain
2Department of Internal Medicine, Ramón y Cajal University Hospital, Spain
*Corresponding author: García-Cosío M Department of Pathology, Ramón y Cajal University Hospital, Carretera de Colmenar KM 9, 100, 28034, Madrid, Spain; CIBERONC, IRYCIS; University of Alcala, Madrid, Spain. Tel: +34-913368140 Email: monica.garciacosio@salud.madrid.org
Received: January 19, 2024 Accepted: February 29, 2024 Published: March 07, 2024
Abstract
Intravascular Large B-Cell Lymphoma (IVLBCL) is an aggressive and rare extranodal B-cell lymphoma characterized by the proliferation of large neoplastic B cells virtually exclusively within the lumen of blood vessels. Three clinical variants have been described: the classic variant, the hemophagocytic syndrome-associated variant and the cutaneous variant. This entity usually is not suspected clinically and the diagnosis is based on histopathological confirmation and in many cases is made at autopsy. We report two autopsy cases of IVLBCL at our institution and a literature review of cases from the last ten years. We also discuss the latest advances about pathogenesis and molecular features of IVLBCL.
Despite advances in understanding the molecular landscape of this disease, earlier diagnosis and treatment remains a challenge.
Keywords: Intravascular large B-cell lymphoma; Review; Diagnosis; Autopsy; Molecular pathology
Background
Intravascular Large B-Cell Lymphoma (IVLBCL), historically known as angiotropic lymphoma, was first described in 1959 as an endothelial neoplasm with vascular spread [1]. Nowadays it is considered a rare subtype of non-Hodgkin lymphoma which has been updated and revised in 2022 World Health Organization (WHO) classification of lymphoid neoplasms. It is defined as an aggressive extranodal B-cell lymphoma characterized by the proliferation of large neoplastic B-cells almost exclusively within the lumen of blood vessels [2]. There are two main histologic variants of IVLBCL, historically classified by geographic presentation: the classic or ‘‘Western’’ type and hemophagocytic syndrome–associated or ‘‘Asian’’ type [3]. However, a third “cutaneous variant” has been described in virtue of their clinical features rather than by their geographical distribution [4]. The classical variant is consistently associated with involvement of systemic organs, whereas the cutaneous variant is restricted to the skin. The hemophagocytic syndrome-associated variant presents as hemophagocytic syndrome, hepatosplenomegaly and thrombocytopenia (>75%) [2]. The latter follows a very aggressive course associated with multiorgan failure, while the cutaneous variant has a better prognosis, possibly related to earlier diagnosis and more favorable clinical features [4,5]. Diagnosis of IVLBCL remains challenging because of the wide variability in clinical presentation. Consequently, it is not uncommon for patients with IVLBCL to have a first diagnosis at post-mortem. Because of the rarity of IVLBCL and the difficulty of its diagnosis, we present two recent autopsy cases and a review of the literature on cases that have been reported in the last ten years.
Case Reports
Case 1
An 87-year-old woman with a previous history of recurrent lower urinary tract infections was admitted to our hospital because of fever and signs of neurological impairment in the last 24 hours. She presented with elevated phase reactants and lower urinary tract infection. She started empirical antibiotic treatment, but she relapsed with fever and clinical worsening. An abdominal CT scan was performed showing a soft tissue mass involving the left middle ureter with extension to the kidney, suggestive of a primary tumor. Other findings were ill-defined liver lesions and pseudonodular thickening of the right adrenal gland. In the following days, the patient presented deterioration with progressive loss of consciousness and marked thrombopenia. A new CT scan showed a reduction in the size of the mass described suggesting an inflammatory lesion. The patient developed unfavorably and died. A clinical autopsy was performed.
Autopsy Findings
Gross findings included mild hepatomegaly (1870 grams; reference range 1500-1800 grams [6]), a congested appearance of the liver and multiple intrahepatic nodules in segment IV. Microscopic examination of several organs showed infiltration by a poorly differentiated, high-grade and discohesive neoplastic cell proliferation. These cells were of medium to large size with sparse cytoplasm and irregular nuclei with vesicular chromatin and prominent nucleolus. Numerous mitotic figures were seen. This proliferation was mainly found in the lumen of small and medium-sized blood vessels (Figure 1). However, we also observed extravascular infiltration in some organs such as the liver and adrenal glands (Figure 2). Additionally prominent histiocytic cell hyperplasia was observed in the spleen and bone marrow, with frequent images of hemophagocytosis (Figure 3). The bone marrow was not infiltrated by lymphoma cells. Immunohistochemistry (Figure 4) showed that the neoplastic cells were positive for CD45, CD20, CD79a, Bcl6 (weak) and CD5 (weak), with negativity for TdT, Bcl2, CD3, CD10, CD21, CD23, CD30, Cyclin-D1, MUM-1 and c-Myc. Epstein-Barr virus was not detected by in situ hybridization (EBER). The tumor was negative for other markers such as CKAE1/AE3, CK7, CK20, CK5/6, S100 and Melan-A. There was no overexpression of p53. No restriction of immunoglobulin light chains (kappa, lambda) was observed. The proliferation index (Ki67) was close to 80%.
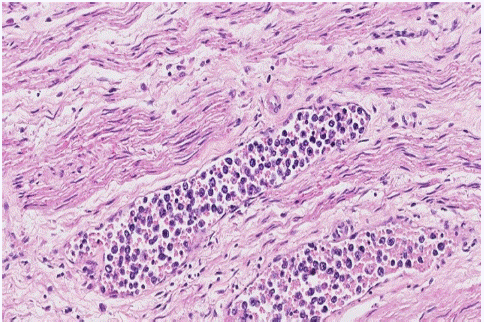
Figure 1: Case 1. In the lumen of medium and small-sized vessels of periureteral soft tissues, a high-grade discohesive neoplastic proliferation with lymphoid appearance was identified. (H&E stain, x20).
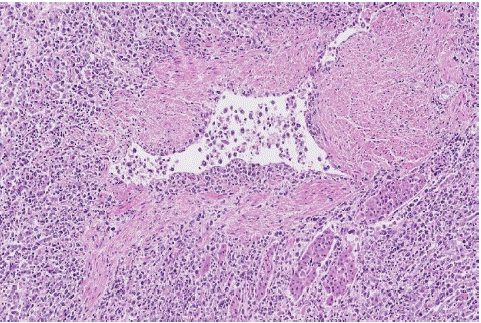
Figure 2: Case 1. Adrenal gland showing extravascular infiltration due to the disruption of the vessel wall with transmural infiltrate and extension into the adjacent parenchyma and interstitium. (H&E stain, x10).
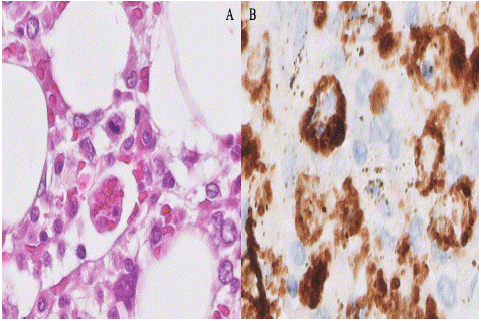
Figure 3: Case 1. Bone marrow. A) Erythrocyte phagocytosis (H&E stain, x60). B) Macrophages with images of hemophagocytosis of nucleated cells highlighted by a CD68 stain (x60).
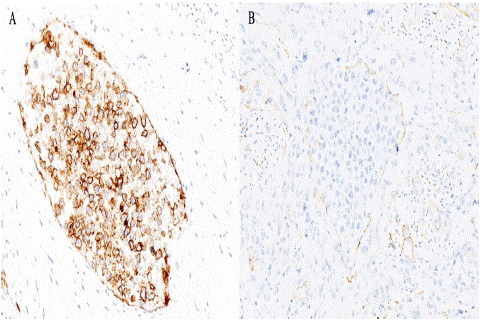
Figure 4: Case 1. Intravascular infiltration of IVLBCL in small and medium sized blood vessels of the myometrium. A) Neoplastic
cellularity positive for CD20 stain (x40). B) CD34 stain enhances the intravascular location of the neoplasm (x40).
As the patient met clinical and analytical criteria for hemophagocytic syndrome, the final diagnosis was IVLBCL associated with hemophagocytic syndrome variant.
Case 2
A 75-year-old man with no relevant medical history was admitted to our hospital with a constitutional syndrome, fever of unknown origin for three weeks and neuropsychiatric symptoms in the last hours. Blood tests revealed thrombopenia and leukopenia. Three possible diagnoses were considered: first, an active tuberculosis infection, which was quickly ruled out; second, IVLBCL, for which two biopsies of normal skin, a liver biopsy and a bone marrow biopsy were performed. The first skin biopsy showed some lymphoid appearing cells within capillaries and vessels of fine caliber. These cells were medium to large and centroblastic in appearance. Immunohistochemistry revealed that the cells were positive for CD20 and BCL2 (weak) and negative for CD10 and CD138 (Figure 5). A definitive diagnosis of IVLBCL could not be made due to the low cellularity represented. The second skin biopsy and the liver biopsy presented no relevant histological lesions. Bone marrow biopsy showed reactive changes and histiocytic cell hyperplasia (CD68 positive) with images of hemophagocytosis without neoplastic infiltrates.
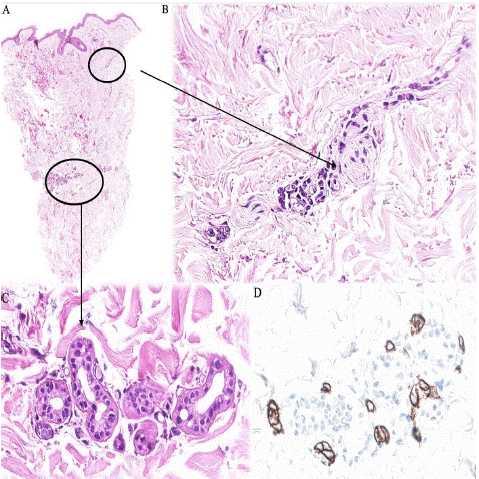
Figure 5: Case 2. Normal skin biopsy. A) There is a preserved epidermis with no significant inflammatory or lymphoid infiltrates in the dermis. (H&E stain, x2). B,C) High power view reveals very few lymphoid-appearing cells inside capillaries and vessels (H&E stain, x40). D) Cellularity is positive with CD20 stain (x40).
The third differential diagnosis was neurolupus, as the patient fulfilled the criteria for this disease, for which he was treated with clinical and laboratory improvement. However, he was readmitted to our center one month later because of persistent fever and neurological deterioration with no response to treatment, so two doses of rituximab were administered. Nevertheless, the patient died and a clinical autopsy was performed.
Autopsy Findings
The main macroscopic findings were the presence of abundant secretions in the upper airways and congested lungs with predominantly apical emphysematous bullae. Brain examination revealed only slightly thinned cerebral gyri and yellowish plaques in the vascular wall of the circle of Willis and the vertebro-basilar system, which did not obstruct the lumen of the vessels. Coronal sections showed 1-2 mm grayish-reddish foci in the periventricular white matter and small cystic lesions in the lenticular nucleus.
The most striking microscopic finding was the presence of a neoplastic, discohesive, medium to large lymphoid cell proliferation (Figure 6), mainly in small and medium caliber vessels of several organs including the brain. The cells had sparse cytoplasm, an irregular nucleus with vesicular chromatin and a prominent nucleolus. Immunohistochemistry showed that these cells were positive for CD79, BCL2, PAX5, MUM1 (+/-) and BCL6 (+/-) and negative for EBER, CD20, CD10 and c-MYC. The CD20 negativity was explained by the doses of rituximab received. The proliferation index (Ki67) was 60%. Bone marrow was hypercellular and revealed signs of hemophagocytosis. These clinical, morphological and immunohistochemical findings were consistent with a multiorgan involvement of IVLBCL, classical variant.
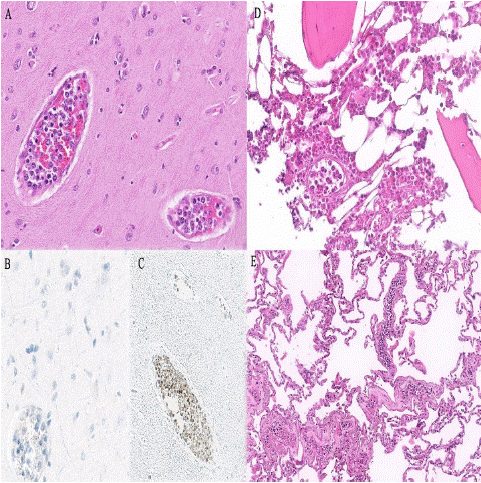
Figure 6: Case 2. A) Lenticular nuclei with several images of neoplastic lymphoid cells inside the small size-vessels (H&E stain, x20). B) The cellularity is negative for CD20 stain (x40). C)The
cellularity is positive for CD79a (x20). D) Bone marrow with
intravascular involvement (H&E stain, x20). E) Lung with
intravascular involvement (H&E stain, x10).
Discussion
IVLBCL is an aggressive and rare extranodal B-cell lymphoma defined by the intravascular location of the neoplasm, which may occasionally be minimally extravascular and can involve virtually any organ [2]. Three clinical variants have been described: the classic variant, which frequently involves the central nervous system and endocrine system; the hemophagocytic syndrome-associated variant, which fulfill the clinical and analytical criteria of hemophagocytic syndrome; and the cutaneous variant with only skin involvement [7]. The prognosis of the first two variants is very poor and the cutaneous variant has a better outcome [2].
Histologically it is defined by highly atypical cells with enlarged, pleomorphic and hyperchromatic nuclei, predominantly located within capillary vessels [8]. On immunohistochemistry they are positive for pan B-cell markers such as CD20 and CD79. Exceptionally, there are cases CD20-negative, where it is necessary to perform other B markers such as CD79a and/or PAX-5 [9]. CD5 is co-expressed in 20-40%. MUM1 is expressed in 75-80%, and most cases have a non-germinal center B-cell immunophenotype, but 13% are CD10-positive [2]. CD34 staining is useful for diagnosis, as it helps to locate the neoplastic cellularity within the vessels [10].
The two cases described above highlight the difficulty in diagnosing IVLBCL because it is a very rare entity with non-specific symptoms. In addition, early diagnosis is challenging with the available diagnostic procedures and in many cases the diagnosis is made at autopsy. We reviewed the literature for case reports of IVLBCL over the last decade (Table 1 [7,8,11–17]), focusing on the time at which the diagnosis was made and the associated clinical course. In the reported cases, there were 5 males and 4 females with a mean age of 63 years. In most cases, patients presented with non-specific symptoms, the most common being fatigue, fever and weight loss. Thrombocytopenia and elevated lactate dehydrogenase were present in almost all cases. Lymphoma was suspected as the primary diagnosis in only two cases and in all cases the diagnosis was made after histological examination. We also observed that 6 out of 9 patients were diagnosed post-mortem and only 3 were diagnosed alive.
![]()
Author (year)
Gender/
Age (onset)Clinical symptoms
Blood test
Primary diagnosis
Biopsy site
IHQ
Outcome and time of diagnosis
[11]
F / 69
Fatigue, weakness, night sweats and fever
Thrombocytopenia, erythrocyte sedimentation, LDH and C-reactive protein elevated
Paraneoplastic syndrome due to lung cancer.
None
CD20+ MUM1+� CD3- CD30-CD15- CD10-BCL6- ALK1-
Died on the third day after admission.
After autopsy.[12]
M / 51
Dry cough and fever
Thrombocytopenia and liver dysfunction
Malignant lymphoma and hemophagocytic syndrome
Bone marrow biopsy
CD20+, CD45+, CD5+, CD31+, BCL-6+, MUM-1+, BCL2+,
CD3-, CD10-.Died 24 days after the onset of the symptoms.
After autopsy.[13]
F / 47
Secondary amenorrhea, fatigue, deteriorating vision and night sweats.
Hyponatremia, anemia and thrombocytopenia. LDH elevated.
Non functionating macroadenoma (pituitary) and malignancy of uncertain primary
Adrenal biopsy
CD20+, BCL2+, CD31+
NA.
Alive.[8]
M / 80
Cough, fatigue, fever, weight loss, and a syncopal episode
Anemia and thrombocytopenia
Infectious process or ehrlichiosis
Bone marrow biopsy
CD20+, CD3+, PAX5+,
CK-Died 4 days after hospital admission.
After autopsy.[14]
F / 51
Cognitive deficits, disorientation, poor short-term memory and truncal ataxia (for the past 2 months)
NA
IVLBCL
Skin biopsy
CD10+, CD20+, BCL2+, BCL6+, MUM1+, PDL1 (50%),
CD3-,
CD5-,
EBER-Died after 3 months of hospitalization.
After autopsy.[7]
F / 66
B symptoms, weight
loss, anemia, and splenomegaly with elevated liver enzymesNA
NA
Bone marrow biopsy
CD20+, PAX5+, CD5+, BCL2+, BCL6+, MUM1+, LEF1+,
CD10-, SOX11-, c-myc-, PD-L1-.NA.
Alive.[15]
M / 50
Paraparesis (myeloradiculitis)
LDH and beta 2 globulin elevated
NA
Bone marrow biopsy and spinal cord biopsy
CD20+, CD45+, CD3+, PDL1+,
CK7-, CK20-, MCK-, TTF1-Thromboembolism and died 2 months after of bilateral pneumonia.
After autopsy.[16]
M / 76
Dysuria
LDH slightly elevated
NA
TURP
CD20+, CD19+, CD5+, MUM1+, c-myc+, BCL2+
CD3-, CD30-, CD10-, BCL6-, Cyclin-D1-, EBER-, EMA-Died 6 months after diagnosis.
Alive.[17]
M / 76
Fatigue, weight loss, altered general
condition and fever. HIV +Lymphopenia, anemia, thrombocytopenia and high ferritin levels
Hantavirus pulmonary syndrome
None
CD20+, CD79+, CD30+, PD-L1+, MUM1+, CD10+, BCL6+, CD138+, EBER+, LMP-1+,
HHV8-,
CD5-,
c-myc-Died 5 days after hospitalization.
After autopsy.
Table 1: Literature review of IVLBCL case reports from the last ten years.
The diagnosis of IVLBCL is also challenging because it does not present as a mass lesion, so it is not easily detected by imaging tests [18]. Nowadays the diagnosis is based on histological confirmation and it often requires multiple biopsies. Indeed, it has been described that random biopsies from normal-appearing skin are highly sensitive compared to bone marrow trephine biopsies [19]. This could be explained because the IVLBCL does not usually involve the bone marrow, nor does it involve lymph nodes, cerebrospinal fluid or peripheral blood [18]. In fact, only 5-10% of IVLBCL exhibit atypical cells on peripheral blood smear [20]. This could be explained by alterations in adhesion molecules required for extravasation (lack of expression of CD29 and CD54, as well as matrix metalloproteinases such as MMP-2 and MMP-9[20]) and chemokines (CXCR3 on lymphocyte and CXCR on endothelial cell [21,22]).
Therefore, a search for new diagnostic methods that allow early detection of IVLBCB is imperative. Currently, liquid biopsy is a non-invasive and promising diagnostic alternative and a useful tool for monitoring residual disease by measuring neoplastic cell-free DNA in plasma (cfDNA). In addition, this technique has allowed a better understanding of the molecular pathogenesis of IVLBCL [23]. The mutational landscape of IVLBCL is similar to that of primary central nervous system lymphoma, primary testicular lymphoma, primary cutaneous Diffuse Large B-Cell Lymphoma (DLBCL) of leg type and a subset of mainly extranodal activated DLBCL [22]. They consist of mutations in molecules involved in B cell/NF- κB receptor signaling (PIM1 [22], MYD88L265P, CD79B) and mutations in SETD1B and HLA-B [23]. Other recurrent mutations have been identified in IRF4, TMEM30A, BTG2 and ETV6, and driver mutations in NOTCH2, CCND3 and GNA13 [22]. A recent analysis of IVLBCL identified mutations in RAC2 due to activation of the BCR pathway [24]. Conventional cytogenetic studies have identified complex karyotypes with recurrent cytogenetic abnormalities involving chromosomes 1, 6q, 8p, 9p, 18, 19q and some samples with tetraploidy [22].
The molecular features of IVLBCL are associated with escape from immune surveillance. Programmed cell Death Ligand 1 (PD-L1) is a cell surface glycoprotein that is aberrantly expressed on neoplastic cells of several tumour types and some lymphomas [25]. PD-L1 interacts with PD-1 on the surface of T cells to modulate the immune response [25]. In a study by Shimada et al [23], they found that 38% of IVLBCL with PD-L1/PD-L2 rearrangements had overexpression of PD-L1/PD-L2. Therefore, those IVLBCL that express PD-L1/PD-L2 may benefit from anti-PD-1 or anti-PD-L1 therapy, which act as immune checkpoint inhibitors. On the other hand, they found alterations in the number of copy genes related to the presentation of antigens to T cells, such as HLA-A/B/C (33%) and HLA class II (9%) [23]. It has been described that PD-L1 is expressed in both the classical and the hemophagocytic variant [25], with heterogeneous expression depending on the anatomical site in which it is located [26].
Classically, treatment of IVLBCL is based on the CHOP regimen (cyclophosphamide, doxorubicin, vincristine and prednisone) [25] as for DLBCL, Not Otherwise Specified (NOS). Following the introduction of rituximab, the prognosis of this disease has improved, with an overall 5-year survival [27]. Nowadays, treatments that target the molecular alterations described above may be a therapeutic key for the future. So far, immune checkpoint inhibitors have shown promising results in some lymphoid malignancies [25].
Conclusion
IVLBCL is an aggressive and rare extranodal B-cell lymphoma. Due to its non-specific and heterogeneous presentation, it is often diagnosed at autopsy. Therefore, this entity should always be considered in the differential diagnosis of elderly patients with fever of unknown origin and/or cytopenias to achieve better survival with the introduction of early treatment. Despite the advances in the molecular landscape of IVLBCL, further research is needed into innovative diagnostic and therapeutic approaches to allow earlier diagnosis and treatment.
References
- Ponzoni M, Ferreri AJM. Intravascular lymphoma: a neoplasm of ‘homeless’ lymphocytes? Hematol Oncol. 2006; 24: 105-12.
- WHO classification of tumours Editorial Board. Haematolymphoid tumours. Lyon (France): International Agency for Research on Cancer; forthcoming. WHO classification of tumours series. 5th ed.11.
- Davis JW, Auerbach A, Crothers BA, Lewin E, Lynch DT, Teschan NJ, et al. Intravascular Large B-Cell Lymphoma. Archives of Pathology & Laboratory Medicine. 2022; 146: 1160-7.
- Ferreri AJM, Campo E, Seymour JF, Willemze R, Ilariucci F, Ambrosetti A, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’1. British Journal of Haematology. 2004; 127: 173-83.
- Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. The Lancet Oncology. 2009; 10: 895-902.
- Finkbeiner WE, Ursell PC, Davis RL. Autopsy pathology: a manual and atlas. 2nd ed. Philadelphia, PA: Saunders/Elsevier. 2009.
- Lindholm KE, Ewalt MD. Hemophagocytic syndrome–associated intravascular large B-cell lymphoma. Blood. 2020; 135: 2432-2432.
- Devitt KA, Gardner JA. Intravascular large B-cell lymphoma: The Great Imitator. Autops Case Rep. 2018; 8: e2018055.
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood. 2018; 132: 1561-7.
- Shimada K, Kiyoi H. Current progress and future perspectives of research on intravascular large B-cell lymphoma. Cancer Science. 2021; 112: 3953-61.
- Thomas CA, Guileyardo JM, Krause JR. An Intravascular Lymphoma with Extravascular Tendencies. Baylor University Medical Center Proceedings. 2014; 27: 341-3.
- Fukunaga H, Kawashima K, Kumakawa H, Hashimoto Y, Takahashi Y. An autopsy of intravascular large B-cell lymphoma with hemophagocytic syndrome. JRSM Open. 2017; 8: 205427041769505.
- Hussain S, Hallam S, Beltran L, Haroon A, Majumdar K, Shamash J, et al. Intravascular large B-cell lymphoma presenting as a pituitary mass with bilateral adrenal enlargement and haemophagocytic lymphohistiocytosis. Br J Haematol. 2018; 181: 851-2.
- Sakakibara A, Inagaki Y, Imaoka E, Ishikawa E, Shimada S, Shimada K, et al. Autopsy case report of intravascular large B-cell lymphoma with neoplastic PD-L1 expression. JCEH. 2018; 58: 32-5.
- Maiese A, La Russa R, De Matteis A, Frati P, Fineschi V. Post-mortem diagnosis of intravascular large B-cell lymphoma. J Int Med Res. 2020; 48: 030006052092426.
- Gu L, Li M, Tong G, Wei W, Fan Y, Zhao Y. Prostate primary intravascular large B-cell lymphoma: A case report. Urology Case Reports. 2022; 45: 102276.
- Veloza L, Tsai CY, Bisig B, Pantet O, Alberio L, Sempoux C, et al. EBV-positive large B-cell lymphoma with an unusual intravascular presentation and associated haemophagocytic syndrome in an HIV-positive patient: report of a case expanding the spectrum of EBV-positive immunodeficiency-associated lymphoproliferative disorders. Virchows Arch. 2022; 480: 699-705.
- Khan MS, McCubbin M, Nand S. Intravascular Large B-Cell Lymphoma: A Difficult Diagnostic Challenge. J Investig Med High Impact Case Rep. 2014; 2: 2324709614526702.
- Matsue K, Asada N, Odawara J, Aoki T, Kimura S ichi, Iwama K ichi, et al. Random skin biopsy and bone marrow biopsy for diagnosis of intravascular large B cell lymphoma. Ann Hematol. abril de 2011; 90: 417-21.
- Charifa A, Paulson N, Levy L, Perincheri S, Lee A, Ko CJ. Intravascular Large B-Cell Lymphoma: Clinical and Histopathologic Findings. Yale J Biol Med. 2020; 93: 35-40.
- Odziomek A, Jarlinski P, Rebizak E, Weglowska J. Intravascular large B-cell lymphoma. pd. 2021; 108: 46-51.
- Gonzalez-Farre B, Ramis-Zaldivar JE, Castrejón de Anta N, Rivas-Delgado A, Nadeu F, Salmeron-Villalobos J, et al. Intravascular Large B-Cell Lymphoma Genomic Profile Is Characterized by Alterations in Genes Regulating NF-κB and Immune Checkpoints. Am J Surg Pathol. 2023; 47: 202-11.
- Shimada K, Yoshida K, Suzuki Y, Iriyama C, Inoue Y, Sanada M, et al. Frequent genetic alterations in immune checkpoint-related genes in intravascular large B-cell lymphoma. Blood. 18 de marzo de 2021; 137: 1491-502.
- Kodgule R, Chen J, Khonde P, Robinson J, D’Albora A, Cook L, et al. Recurrent switch 2 domain RAC2 mutations in intravascular large B-cell lymphoma. Blood Adv. 2022; 6: 6051-5.
- Gupta GK, Jaffe ES, Pittaluga S. A study of PD-L1 expression in intravascular large B cell lymphoma: correlation with clinical and pathological features. Histopathology. 2019; 75: 282-6.
- Sakakibara A, Inagaki Y, Imaoka E, Sakai Y, Ito M, Ishikawa E, et al. Divergence and heterogeneity of neoplastic PD-L1 expression: Two autopsy case reports of intravascular large B-cell lymphoma. Pathology International. 2019; 69: 148-54.
- Rajyaguru DJ, Bhaskar C, Borgert AJ, Smith A, Parsons B. Intravascular large B-cell lymphoma in the United States (US): a population-based study using Surveillance, Epidemiology, and End Results program and National Cancer Database. Leuk Lymphoma. 2017; 58: 1-9.