
Special Article: Prostate Cancer
Ann Hematol Onco. 2024; 11(2): 1451.
Lineage Plasticity in Prostate Cancer: A Review of Recent Advances
Xiaoyan Qiang¹; Yucheng Zeng¹; Edison Wu²; Peng Peng¹*
1TransThera Sciences (Nanjing), Inc., Fl 3, Bld 9, Phase 2 Accelerator, Biotech and Pharmaceutical Valley, The People’s Republic of China
2RGS Guilford Nanjing, No.17, Kangjian Road, Jiangbei New Area, Nanjing
*Corresponding author: Peng Peng Address: TransThera Sciences (Nanjing), Inc., Fl 3, Bld 9, Phase 2 Accelerator, Biotech and Pharmaceutical Valley, Jiangbei New Area, Nanjing, Jiangsu, 210032, The People’s Republic of China. Email: peng_peng@transtherabio.com
Received: April 01, 2024 Accepted: May 02, 2024 Published: May 09, 2024
Abstract
Lineage plasticity, the capacity of cancer cells to transition between different cell states, has emerged as a critical mechanism of therapy resistance in prostate cancer. This review summarizes recent insights into the molecular drivers, clinical implications, and potential therapeutic targets associated with lineage plasticity in prostate cancer, focusing on the JAK/STAT, FGFR, and KIT signaling pathways.
Introduction
Prostate Cancer (PCa) ranks as the most diagnosed tumor and the second leading cause of cancer-related deaths among men in the US, comprising an estimated 29% of new diagnoses and 11% of total cancer-related morbidity in 2024 [1]. While Androgen Receptor (AR) signaling plays a pivotal role in PCa development, Androgen Deprivation Therapy (ADT) followed by AR Signaling Inhibitors (ARSIs) has been the cornerstone of treatment [2-5]. However, despite advancements with ARSIs, therapy duration remains limited due to emerging resistance mechanisms, including AR-dependent mutations, Amplification and Splice Variants (AR-Vs); AR bypass such as Glucocorticosteroid Receptor (GR), and other AR-independent mechanisms. Among these, lineage plasticity, the ability of cancer cells to transition between different cell states, has been proposed to play a critical role in hormone therapy resistance in PCa [6,7].
Lineage plasticity in prostate cancer, characterized by shifts from Androgen Receptor (AR)-positive adenocarcinoma to AR-low or AR-null Neuroendocrine Prostate Cancer (NEPC), has emerged as a hallmark of Castration-Resistant Prostate Cancer (CRPC) and a significant therapeutic challenge [2,3,4,7]. This review explores the molecular mechanisms driving lineage plasticity, with particular emphasis on recent advances post-2022.
Characteristics of Lineage Plasticity in PCa Progression
ADT is still the mainstay treatment of newly diagnosed patients, and new generation of ARSIs are well-developed to blunt the post-ADT androgen synthesized intratumorally or metabolized by adrenally-produced androgen precursors [8]. In the past decade, novel ARSIs interfering AD/androgen interaction – such as enzalutamide, apalutamide and darolutamide- or blocking androgen biosynthesis – for instance abiraterone acetate and ODM-208 [9] – have been well-studied (or still ongoing) and approved (or in late clinical stage) for the treatment of CRPC. However, post-ARSI resistance is emerging recently, among which approximately 20% of CRPC tumors represent NEPC features [10].
The recent research has elucidated the molecular events occurring in early-stage prostate cancer cells that confer the capability of lineage plasticity. Loss-of-function mutations in tumor suppressor genes RB1, TP53, and PTEN, alone or in combination, contribute significantly to lineage plasticity. The comprehensive genomic analysis of NEPC found that the concomitant loss of more than one of these critical factors is enriched in NEPC other than PCa. For instance, RB1 loss and the mutation or deletion of TP53 occur together more commonly in NEPC tumors (53.3%) than in castration-resistant adenocarcinoma PCa (13.7%) [11]. Ku’s study [12] suggests that Rb1 loss plus Pten mutation facilitates lineage plasticity and metastasis of prostate adenocarcinoma, while with additional loss of Trp53 causes resistance to ADT. TP53 and RB1 suppress the expression of epigenetic reprogramming factors such as Ezh2, Sox2, and Sox9, and upregulation of these epigenetic reprogramming factors in the genetic background of TP53 and/or RB1 loss leads to the establishment of a pluripotent stem-like environment that induces plastic status with multiple lineage potentials in prostate cancer cells [11,13-15]. Additionally, amplifications of MYCN and AURKA also contribute to the genome-wide rewiring. Therefore, lineage plasticity, stemming from the loss of tumor suppressor genes, represents an inherent characteristic of cancer (a cell-autonomous process), and its progression is expedited by ARSIs treatment and modulated by the tumor microenvironment.
In line with the concept of cell-autonomy, although still subject to debate, evidence indicates a transdifferentiation process from the adenocarcinoma lineage to the neuroendocrine lineage, rather than the clonal selection of pre-existing cells with alternative lineages. Support for this transdifferentiation hypothesis comes from findings that luminal-specific genomic alterations, such as ERG translocation, are retained in Neuroendocrine Prostate Cancer (NEPC) [2,8,16].
Lineage plasticity is characterized by a gradual decline in AR expression and luminal markers, accompanied by the acquisition of basal and Neuroendocrine Prostate Cancer (NEPC) markers. Throughout this process, Prostate Cancer (PCa) displays intermediate states with a blend of basal, luminal, Epithelial-Mesenchymal Transition (EMT), stemness, and NEPC features (Figure 1). Plasticity is first initiated in an epithelial population defined by mixed luminal-basal phenotype [2]. NEPC tumors, the end stage of lineage plasticity, undergo a histological transformation marked by elevated expression of neuroendocrine markers and decreased expression of luminal markers, indicating a shift away from AR signaling dependency [8]. Understanding the molecular characteristics and driving pathways of lineage plasticity at different stages may provide unique opportunities for pharmacological intervention to prevent, delay, or even reverse plasticity.
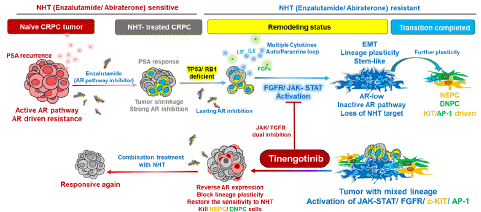
Figure 1: The process of lineage remodeling and treatment strategy in CRPC.
For naive Castration-Resistant Prostate Cancer (CRPC) relapsed after Androgen Deprivation Therapy (ADT) with increased prostate specific antigen (PSA, also known as KLK3) level, initial responses to Novel Hormonal Therapy (NHT) are marked by the PSA reduction, indicating tumor regression. However, prolonged androgen receptor (AR) suppression and genetic alterations (especially TP53/Rb1 loss) can lead to lineage plasticity, resulting in heterogeneous, NHT-resistant cancer cells. This phenotypic remodeling involves activation of the JAK-STAT and FGFR pathways, driving cells into a pluripotent state with the potential to differentiate into various subtypes. This stage is characterized by EMT and increased stemness, with a downregulation of luminal markers and AR signaling. Dual inhibition of JAK-STAT and FGFR can potentially reverse this remodeling process in transitional cells, reverting them to AR-dependent states and restoring NHT sensitivity. After the transition phase, the lineage remodeling is irreversible through the blockade of JAK and FGFR pathways. Cells manifest as heterogeneous tumors with distinct subtypes, such as NEPC and DNPC, each driven by specific oncogenic pathways like KIT, AP-1 or WNT. However, it’s promising to eliminate these cells through targeting the specific driver pathways, such as KIT or AP-1 upstream signaling.
Tinengotinib, a multi-kinase inhibitor, exhibiting great inhibition on JAKs, FGFRs, Aurora kinases and KIT. Through blocking NE-lineage remodeling to restore the sensitivity of NHT, tinengotinib combined with NHT possesses great potential in treating heavily-treated CRPC.
The intermediate lineage each expresses a distinct group of biomarkers. NEPC lineage relinquishes reliance on AR signaling and adopts a histological profile of neuroendocrine differentiation. Generally, NEPC tumors exhibit positivity for Numerous Neuroendocrine (NE) markers, including Synaptophysin (SYP), CD56 (NCAM), Chromogranin A/B (CHGA/B), neuron-specific enolase (NSE), INSM1, and ASCL1, while typically lacking expression of luminal markers like PSA, which is closely associated with the AR signaling pathway [8]. Furthermore, the intermediate lineages observed in the progression of Castration-Resistant Prostate Cancer (CRPC) extends beyond NEPC, encompassing other lineage signatures such as increased EMT and pluripotent stemness [8,11,13-15]. Typical markers for each lineage characteristic are detailed in Table 1.
![]()
Representative Markers
Luminal and AR Pathway Markers
AR, KLK3 (PSA), KRT8, KRT18, NKX3-1, FOXA1 TMPRSS2, HOXB13, FOLH1, DPP4, TFF3
Basal Markers
TP63, KRT5, KRT14
NE Markers
ENO2, NCAM1, CHGA, SYP, CHGB
NE Transcriptional Factors
POU3F2, ASCL1, NEUROD1, INSM1
Stem and Pluripotent Transcriptional Factors
SOX2, POU5F1, NANOG, PAX6
Table 1: Representative markers for each lineage.
Molecular Mechanisms Driving Lineage Plasticity in PCa Progression
JAK/STAT/IL-6 axis
The JAK/STAT/IL-6 signaling pathway is a complex cellular communication system that plays a crucial role in various biological processes, including immune responses, cell growth, differentiation, and survival. Recent research on the JAK/STAT/IL signaling pathway in prostate cancer lineage plasticity has provided significant insights into the mechanisms that drive the progression and resistance to therapy (especially ADT/ARSIs) in prostate cancer.
Deng et al. [17] reveals that the ectopic (abnormal) activation of JAK-STAT signaling is required for lineage plasticity-driven resistance to AR-targeted therapy in metastasis CRPC, particularly in the context of TP53/RB1 deficiency and SOX2 upregulation. This activation allows cancer cells to transition to a stem-like and multilineage state, which confers resistance to therapy. The data showed that JAK-STAT signaling is specifically required for subclones expressing stem-like and multilineage transcriptional programs, rather than those exclusively expressing a neuroendocrine-like lineage program. Both genetic and pharmaceutical inhibition of key components of the JAK-STAT pathway, including JAK1/JAK2 and STAT1/STAT3, can resensitize resistant mCRPC cells to AR-targeted therapy.
Also, in 2022, Chan et al. [2] published their investigations on the role of JAK/STAT inflammatory signaling in lineage plasticity in prostate cancer on science. They observed that the loss of tumor suppressor genes Trp53 and Rb1 led to the lineage transitions, depending not only on elevated JAK activity, but also FGFR. With combinatorial inhibition of JAK and FGFR pathways led to the reversion to a more luminal phenotype and increased sensitivity to the anti-androgen drug enzalutamide, indicating dual inhibition of JAK and FGFR pathways could be a potential therapeutic strategy to overcome this resistance and improve treatment outcomes for patients with CRPC by resensitizing end-stage CRPC to ARSIs with AR recovery.
FGFR
FGFR, or fibroblast growth factor receptor, is a family of Receptor Tyrosine Kinases (RTKs) that is expressed on the cell membrane and play crucial roles in various cellular functions, including migration, proliferation, differentiation, and survival. FGFR signaling pathway is significantly activated in certain molecular subtypes of CRPC, and plays a significant role in the biological characteristics of prostate stem cells, particularly in maintaining the stem cell state and promoting their differentiation, indicating its great potential to treat end-stage CRPC patients [18-21].
Huang’s lab [18,19] report that prostaspheres, a Three-Dimensional (3D) spherical valuable model for studying the properties of prostate stem cells, are derived from the P63-expressing P-bSCs (basal prostate stem cells), which contain both quiescent and cycling cells that can either differentiate to basal epithelial or luminal epithelial cells. FGF/FGFR signaling is required for self-renewal and stemness of P-bSCs, as treatment of cells with FGF7 or FGF10 increases sphere formation and ablation of FGFR2 or FRS2 in P-bSCs abrogates such elevation. Juyeon et al. [20] also provide evidence that FGFR1 plays an essential role in the proliferation of PCa CSCs (cancer stem cells) at both molecular and cellular levels.
Estefania et al. [22] demonstrates that FGFR1 and its isoforms have a significant impact on the metastatic progression of PCa, particularly to bone. The increased expression of FGFR1 in CRPC bone metastases and its association with genes like LAD1 highlight its role in facilitating the spread of cancer cells to the bone. FGFR is regulated in prostate cancer through various mechanisms, including the promotion of its expression and activation by proteins like ID2 [23] and Gremlin1 [24], as well as by circular RNAs such as circFGFR1int2 [25]. These regulators contribute to the progression of the disease and the development of resistance to treatments like ADT and ARSIs.
FOXA2/cKIT
Ming et al. [26] identifies FOXA2 as a pioneer factor that orchestrates the adeno-to-neuroendocrine lineage transition in prostate cancer. FOXA2 expression is significantly induced by ADT, and its knockdown can reverse the transition from adenocarcinoma to neuroendocrine cancer, while FOXA1 has been extensively studied for its role in maintaining the luminal phenotype. FOXA2 directly promotes the Kit expression, and this activation of the KIT pathway is specific to neuroendocrine cells and plays a crucial role in maintaining their proliferation. Pharmacological inhibition of the KIT pathway by imatinib, a KIT inhibitor, significantly suppresses tumor growth in both mouse and human NEPC models, indicating the potential of KIT inhibitors in treating CRPC patients in the future.
Conclusions and Therapeutic Implications
Prostate cancer progression, driven by lineage plasticity, entails a shift from androgen-dependent to androgen-independent states. Lineage plasticity poses a complex and multifaceted challenge in prostate cancer, complicating late-stage outcomes in Castration-Resistant Prostate Cancer (CRPC) patients, particularly those who develop resistance to Androgen Receptor Signaling Inhibitors (ARSIs). End-stage CRPC is historically marked by phenotypic heterogeneity, making it difficult to definitively classify tumors as entirely AR-positive or NEPC in black and white.
Comprehending the molecular mechanisms underlying lineage plasticity is crucial for devising novel therapeutic strategies. The identification of key molecular drivers such as the JAK/STAT, FGFR, and cKIT signaling pathways, alongside the clinical characterization of NEPC, has facilitated the development of targeted therapies. Continuous research into lineage plasticity mechanisms and the exploration of innovative therapeutic approaches aimed at impeding or reversing lineage transitions are imperative for enhancing outcomes in patients with advanced prostate cancer.
The field of prostate cancer research is advancing rapidly. Future studies should prioritize the development of robust preclinical in vitro and in vivo models, the exploration of biomarkers for lineage plasticity, the identification of novel therapeutic targets, and the design of clinical trials to assess the efficacy of targeted therapies. Tinengotinib, the sole kinase inhibitor targeting all aforementioned pathways—JAK, FGFRs, and Aurora kinases—has demonstrated promising potential in treating advanced-stage CRPC [27]. As of August 28, 2023, in a published study, 22 out of 30 heavily treated metastatic CRPC patients were assessable for efficacy, with 13 evaluated using RECIST v1.1 criteria and 14 assessed based on PSA levels. The median Radiographic Progression-Free Survival (rPFS), Overall Response Rate (ORR), and Disease Control Rate (DCR) were 5.6 months, 46% (6/13), and 85% (11/13), respectively, while the PSA50 response rate was 50% (7/14). With a deeper understanding of the mechanisms governing lineage plasticity, it is expected that more effective personalized treatment strategies will emerge, leading to improved patient outcomes.
References
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA: a cancer journal for clinicians. 2024; 74: 12–49.
- Chan JM, Zaidi S, Love JR, Zhao JL, Setty M, Wadosky KM, et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science (New York, N.Y.). 2022; 377: 1180–1191.
- Kotamarti S, Armstrong AJ, Polascik TJ, Moul JW. Molecular Mechanisms of Castrate-Resistant Prostate Cancer. The Urologic clinics of North America. 2022; 49: 615–626.
- Blee AM, Huang H. Lineage plasticity-mediated therapy resistance in prostate cancer. Asian journal of andrology. 2019; 21: 241–248.
- Buttigliero C, Tucci M, Bertaglia V, Vignani F, Bironzo P, Di Maio M, et al. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer treatment reviews. 2015; 41: 884–892.
- Wang Y, Chen J, Wu Z, Ding W, Gao S, Gao Y, et al. Mechanisms of enzalutamide resistance in castration-resistant prostate cancer and therapeutic strategies to overcome it. British journal of pharmacology. 2021; 178: 239–261.
- Imamura J, Ganguly S, Muskara A, Liao RS, Nguyen JK, Weight C, et al. Lineage plasticity and treatment resistance in prostate cancer: the intersection of genetics, epigenetics, and evolution. Front. Endocrinol. 2023; 14: 1191311.
- Storck WK, May AM, Westbrook TC, Duan Z, Morrissey C, Yates JA, et al. The Role of Epigenetic Change in Therapy-Induced Neuroendocrine Prostate Cancer Lineage Plasticity. Frontiers in endocrinology. 2022; 13: 926585.
- Karimaa M, Riikonen R, Kettunen H, Taavitsainen P, Ramela M, Chrusciel M, et al. First-in-Class Small Molecule to Inhibit CYP11A1 and Steroid Hormone Biosynthesis. Molecular cancer therapeutics. 2022; 21: 1765–1776.
- Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2018; 36: 2492–2503.
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016; 22: 298–305.
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (New York, N.Y.). 2017; 355: 78–83.
- Ma M, Zhu Y, Xiao C, Li R, Cao X, Kang R, et al. Novel insights into RB1 in prostate cancer lineage plasticity and drug resistance. Tumori. Advance online publication. 2024.
- Nouri M, Massah S, Caradec J, Lubik AA, Li N, Truong S, et al. Transient Sox9 Expression Facilitates Resistance to Androgen-Targeted Therapy in Prostate Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2020; 26: 1678–1689.
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science (New York, N.Y.). 2017; 355: 84–88.
- Beltran H, Demichelis F. Therapy considerations in neuroendocrine prostate cancer: what next?. Endocrine-related cancer. 2021; 28: T67–T78.
- Deng S, Wang C, Wang Y, Xu Y, Li X, Johnson NA, et al. Ectopic JAK–STAT activation enables the transition to a stem-like and multilineage state conferring AR-targeted therapy resistance. Nat Cancer. 2022; 3: 1071–1087.
- Huang Y, Hamana T, Liu J, Wang C, An L, You P, et al. Prostate Sphere-forming Stem Cells Are Derived from the P63-expressing Basal Compartment. The Journal of biological chemistry. 2015; 290: 17745–17752.
- Huang Y, Hamana T, Liu J, Wang C, An L, You P, et al. Type 2 Fibroblast Growth Factor Receptor Signaling Preserves Stemness and Prevents Differentiation of Prostate Stem Cells from the Basal Compartment. The Journal of biological chemistry. 2015; 290: 17753–17761.
- Ko J, Meyer AN, Haas M, Donoghue DJ. Characterization of FGFR signaling in prostate cancer stem cells and inhibition via TKI treatment. Oncotarget. 2021; 12: 22–36.
- Labrecque MP, Brown LG, Coleman IM, Nguyen HM, Dalrymple S, Brennen WN, et al. Targeting the fibroblast growth factor pathway in molecular subtypes of castration-resistant prostate cancer. The Prostate. 2024; 84: 100–110.
- Labanca E, Yang J, Shepherd PDA, Wan X, Starbuck MW, Guerra LD, et al. Fibroblast Growth Factor Receptor 1 Drives the Metastatic Progression of Prostate Cancer. European urology oncology. 2022; 5: 164–175.
- Zhang J, Chen Z, Mao Y, He Y, Wu X, Wu J, et al. ID2 Promotes Lineage Transition of Prostate Cancer through FGFR and JAK-STAT Signaling. Cancers. 2024; 16: 392.
- Cheng C, Wang J, Xu P, Zhang K, Xin Z, Zhao H, et al. Gremlin1 is a therapeutically targetable FGFR1 ligand that regulates lineage plasticity and castration resistance in prostate cancer. Nature cancer. 2022; 3: 565–580.
- Wang R, Zhong J, Pan X, Su Z, Xu Y, Zhang M, et al. A novel intronic circular RNA circFGFR1int2 up-regulates FGFR1 by recruiting transcriptional activators P65/FUS and suppressing miR-4687-5p to promote prostate cancer progression. Journal of translational medicine. 2023; 21: 840.
- Han M, Li F, Zhang Y, Dai P, He J, Li Y, et al. FOXA2 drives lineage plasticity and KIT pathway activation in neuroendocrine prostate cancer. Cancer cell. 2022; 40: 1306–1323.e8.
- Sarina A Piha-Paul, Sanjay Goel, Chih-Yi Liao, Nashat Y Gabrail, Farshid Dayyani, Syed Mohammad Ali Kazmi, et al. Preliminary safety and efficacy of tinengotinib tablets as monotherapy and combination therapy in advanced solid tumors: A phase Ib/II clinical trial. Journal of Clinical Oncology. 2023; 41.