
Case Report
Ann Hematol Oncol. 2025; 12(3): 1481.
Anemia and End-Stage Renal Disease: How a Wealth of Wisdom (And Crystals) Led to an Unusual Diagnosis
Bardhan R1*, Patel A2 and Sharma D3,4
1Department of Internal Medicine, The Jewish Hospital-Mercy Health, USA
2Florida Cancer Specialist, USA
3Division of Hematology-Oncology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
4Division of Transfusion Medicine, Department of Pathology, Microbiology & Immunology, Vanderbilt University Medical Center, Nashville, TN, USA
*Corresponding author: Bardhan R, Department of Internal Medicine, The Jewish Hospital- Mercy Health, 4777 E Galbraith Rd, Cincinnati, OH 45236, USA Tel: 408-375-3828; Email: rbardhan@mercy.com
Received: May 07, 2025 Accepted: May 27, 2025 Published: May 30, 2025
Abstract
Hyperoxaluria is a rare disease state characterized by increased amounts of oxalate depositing in the kidneys. The causes of hyperoxaluria can be further divided into primary hyperoxaluria, where there is a genetic enzymatic deficiency leading to an overproduction of oxalate and secondary hyperoxaluria, where either intestinal disorders or increased dietary forms of oxalate are consumed, leading to increased oxalate concentrations. Hyperoxalosis occurs when concentrations of calcium oxalate exceed the renal excretion capacity, leading to systemic deposits in various organs from increased oxalate in the blood. We report a case of a young male with end-stage renal disease (ESRD), recurrent nephrolithiasis, medullary sponge kidney, and transfusion-dependent anemia. Peripheral smear revealed dacrocytes, and bone marrow biopsy showed oxalate crystals consistent with primary oxalosis contributing to his transfusiondependent anemia with suboptimal response to erythropoietin stimulating agents. Molecular testing confirmed primary hyperoxaluria type 1 (PH1) due to AGXT mutations. He was treated with hydration, high-dose pyridoxine, and monitored for nephrolithiasis. This case highlights the importance of considering primary hyperoxaluria in patients with impaired renal function and non-diagnostic anemia work up.
Keywords: Hyperoxaluria; Primary hyperoxalosis; Anemia; Myelophthisic anemia; Oxalate crystals; Oxalate biosynthesis
Abbreviations
AGT: Alanine-Glyoxylate Aminotransferase; AGXT: Alanine- Glyoxylate Aminotransferase Gene; EBV: Epstein-Barr Virus; ESRD: End-Stage Renal Disease; HAO1: Hydroxyacid Oxidase 1; HIV: Human Immunodeficiency Virus; MCV: Mean Corpuscular Volume; OMIM: Online Mendelian Inheritance In Man; PH: Primary Hyperoxaluria; PH1: Primary Hyperoxaluria Type 1.
Introduction
Oxalate is a carbon “waste” metabolite as a byproduct of cellular metabolism. Ex vivo sources of oxalate come from diet, such as fruits, nuts, grains and vegetables. Hyperoxaluria is characterized by a group of rare diseases in which cellular concentrations of oxalate exceeds normal capacity, thereby leading to excretion in the urine. It can be further classified into primary and secondary hyperoxaluria [1]..
Primary hyperoxaluria (PH) is caused by an enzymatic deficiency that leads to overproduction of oxalate. As a result, high amounts of oxalate deposits on the kidney and large quantities of oxalate is found in the urine [1]. In contrast, secondary hyperoxaluria occurs from increased ingestion of dietary oxalate, precursors of oxalate or alterations in intestinal flora [1].
The prevalence of this disease ranges from 1 to 3 per million in the United States with an approximate incidence rate of 1 in 58,000 worldwide [1]. The estimated overall incidence continues to climb over time, which can be attributed to cultural, genetic and dietary concerns. Hyperoxaluria is found in about 25%-45% of all patients who are prone to recurrent calcium kidney stones [1].
Case Presentation
A 28 year-old African American male with a history of recurrent nephrolithiasis and ESRD on hemodialysis, secondary to medullary sponge kidney, presented to the clinic for evaluation of transfusiondependent normocytic normochromic anemia, despite being on an erythropoietin stimulating agent for two months prior to the consultation.
On physical examination and review of systems, there was no lymphadenopathy, hepatosplenomegaly or joint deformities. Blood work revealed normocytic anemia, MCV of 83.3, reticulocyte count <1% and a hemoglobin of 7.0 g/dL. Further workup of anemia was negative, including nutritional deficiencies, hemolysis labs, HIV, Parvovirus B19, and EBV. Peripheral smear showed an abundance of dacrocytes, concerning for myelophthisis (Figure A).With no clear etiology of anemia while requiring one to two transfusions a week, a bone marrow biopsy was performed to evaluate for an aplastic process. Bone marrow biopsy showed infiltrative oxalate crystals (Figure B and Figure C) suggestive of hyperoxalosis under near polarization, raising suspicion for metabolic disease (Figure D). Molecular testing confirmed two different heterozygous variants in the AGXT gene (chromosome 2q37.3) gene (OMIM:604285) with a 50% variant allele frequency, confirming the diagnosis of primary hyperoxaluria. These variants were identified on separate chromosomes, indicating biparental inheritance.
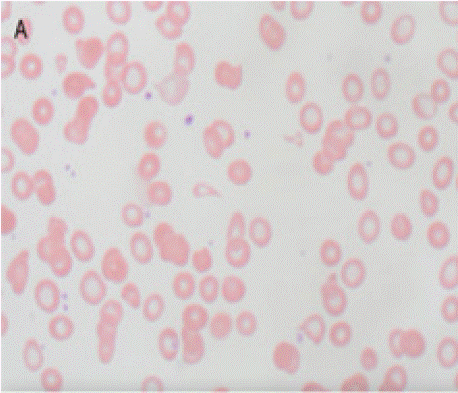
Figure A: Wright stained peripheral blood demonstrating dacrocytes
(teardrop cells), a common feature seen with fibrotic bone marrow.

Figure B: Infiltrative oxalate crystals seen in bone marrow.
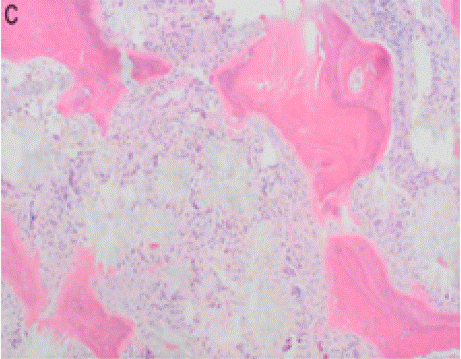
Figure C: Crystal occupying the marrow space and highlighting thickened
bone trabeculae.

Figure D: Morphologic features of the oxalate crystals in the bone marrow,
demonstrated under polarized light.
Following this diagnosis of primary hyperoxalosis, our patient was subsequently initiated on high-dose pyridoxine with hydration to mitigate the production of oxalate crystals, with continuation of erythropoietin stimulating agents, transfusions, and periodic monitoring for nephrolithiasis. Unfortunately, this patient continued to be transfusion dependent, leading to discussions about kidney-liver transplantation to correct the enzymatic defect causing hyperoxaluria and myelophthisic anemia.
Discussion
In primary hyperoxalosis, oxalate is overproduced by the liver. Initially, the kidneys are affected due to calcium oxalate deposition, leading to kidney stone formation and nephrocalcinosis. Over time, the kidney function declines, leading to oxalosis in other different organs.
There are primarily three distinct subtypes of primary hyperoxaluria, which are further categorized based on the different enzymes that are affected in the glyoxylate metabolism pathway. AGXT is expressed in the liver and provides the backbone in the production of the alanine-glyoxylate-aminotransferase (AGT) gene. In type 1 (PH1), glycolate is converted to glyoxylate by glycolate oxidase, which is further metabolized by alanine-glyoxylate-aminotransferase (AGT) to glycine. When there is a deficiency in AGT, glyoxylate accumulates in the cytosol and is converted to oxalate by lactate dehydrogenase. AGT deficiency has been linked to the AGXT gene mutation [1,2,3].
Primary hyperoxalosis is an uncommon and rare diagnosis, typically only recognized after the onset of ESRD and the need for dialysis. Patients often have a 24-hour urinary oxalate level greater than 75 mg, with many exceeding 100 mg or more per day [1].
Primary hyperoxalosis, an autosomal recessive condition, is characterized by oxalate crystal deposition in various organs. This occurs when concentrations of calcium oxalate exceed the renal excretion capacity, leading to systemic crystal deposits in various organs. This condition is progressive, but severity is dependent on the degree of penetrance [4]. Oxalate accumulation in the bone marrow as a cause of myelophthisic anemia is a rare phenomenon, with only limited cases reported in the medical literature [5]. Treatment for primary hyperoxalosis is individualized and includes a low-oxalate diet, increased hydration and medications to prevent kidney stones, such as potassium citrate. In severe cases, kidney and liver transplants may be considered. Combined liver-kidney transplantations are associated with the best outcomes observed as it addresses the underlying enzymatic defect [2,6]. Newer therapies approved for primary hyperoxaluria type 1, such as Lumasiran and Nedosiran, aim to reduce oxalate production by blocking the HAO1 gene in the liver, reducing the production of glycolate oxidase and lowering oxalate levels in primary hyperoxaluria type 1 [6,7].
Oxalate crystal deposition in the bone marrow represents an underrecognized cause of anemia, particularly in patients with renal failure. Initiation of therapies to reduce oxalate production may improve anemia and reduce transfusion dependence in affected individuals. In patients with end-stage renal disease, combined liverkidney transplantation has yielded the most favorable outcomes, as it effectively corrects the underlying enzyme deficiency [2].
Key Points
1. Hyperoxaluria is a condition characterized by the overproduction of oxalate, resulting in the accumulation of oxalate crystals in the kidneys, which can cause kidney damage. Hyperoxalosis is an autosomal recessive condition characterized by oxalate crystal deposition within various organs.
2. Excess oxalate deposits lead to kidney stones, nephrocalcinosis, and progressive organ damage, as seen in the patient’s case of oxalosis. Reports of oxalate deposition in the bone marrow causing myelophthisic anemia are exceedingly rare and are sparsely reported in medical literature.
3. There are two types of hyperoxaluria. Primary hyperoxaluria is caused by genetic enzymatic deficiencies, and secondary hyperoxaluria is due to dietary factors or intestinal disorders. Primary hyperoxaluria is further divided into 3 subtypes, categorized on the different enzymes that affect glyoxylate metabolism pathway.
4. Diagnosis is based on clinical symptoms, urine testing for oxalate levels, and genetic testing, with primary hyperoxaluria confirmed through mutations in genes like AGXT.
5. Treatment focuses on supportive care, such as hydration, dialysis, pyridoxine (B6) and medications to prevent kidney stones and other end-organ complications, such as myelophthisic anemia. Organ transplantation may be required in severe cases to correct the underlying enzymatic deficiency causing hyperoxaluria. Recently approved therapies aim to reduce oxalate production through targeting the HAO1 gene, which is responsible for oxalate production.
References
- Shah A, Leslie SW, Ramakrishnan S. Hyperoxaluria. [Updated 2024 Mar 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025.
- Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol. 2011; 2011: 864580.
- Lorenzo V, Torres A, Salido E. Primary hyperoxaluria. Nefrologia. 2014; 34: 398-412.
- Hopp K, Cogal AG, Bergstralh EJ, Seide BM, Olson JB, Meek AM, et al. Rare Kidney Stone Consortium. Phenotype-Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. J Am Soc Nephrol. 2015; 26: 2559-2570.
- McKenna RW, Dehner LP. Oxalosis. An unusual cause of myelophthisis in childhood. Am J Clin Pathol. 1976; 66: 991-997.
- Dejban P, Lieske JC. New therapeutics for primary hyperoxaluria type 1. Curr Opin Nephrol Hypertens. 2022; 31: 344-350.
- Baum MA, Langman C, Cochat P, Lieske JC, Moochhala SH, Hamamoto S, et al. PHYOX2 study investigators. PHYOX2: a pivotal randomized study of nedosiran in primary hyperoxaluria type 1 or 2. Kidney Int. 2023; 103: 207- 217.
- Kim MJ, Park PW, Seo YH, Kim KH, Seo JY, Jeong JH, et al. Bone Marrow Oxalosis in a Patient With Pancytopenia Following Bilateral Nephrectomy. Ann Lab Med. 2016; 36: 266-267.