
Special Article - Multiple Myeloma
Ann Hematol Oncol. 2015;2(2): 1025.
Drug Resistance in Multiple Myeloma: How to Cross the Border
Manoj K Pandey1, Shantu G Amin1, Maurizio Zangari2 and Giampaolo Talamo1*
1Department of Medicine, Pennsylvania State University College of Medicine, USA
2Myeloma Institute for Research and Therapy, University of Arkansas for Medical Science, USA
*Corresponding author: Giampaolo Talamo, Department of Medicine, Pennsylvania State University College of Medicine, Hershey, Pennsylvania, USA 17033
Received: December 15, 2014; Accepted: January 27, 2015; Published: January 29,2015
Abstract
Multiple myeloma is a hematological malignancy of plasma cells, characterized by a high level of genetic instability, with various gene mutations and chromosomal translocations. Despite the introduction of effective novel agents in the treatment of myeloma, i.e., proteasome inhibitors and Immunomodulatory Drugs (IMiDs), disease relapse and progression occur almost universally, due to innate and acquired drug resistance. We here describe molecular mechanisms and pathways of resistance so far elucidated. Due to the genetic complexity of myeloma, characterized by intra-clonal heterogeneity and progressing with branching evolutionary patterns, we believe that a single general strategy to overcome drug resistance in all patients cannot be developed. However, it is possible that in the future drug resistance will be prevented or treated with the individualized application of genomic and proteomic analyses, targeting the vulnerable pathways of a specific patient in a specific phase of the disease.
Keywords: Drug resistance; Multiple myeloma; Thalidomide; Lenalidomide; Pomalidomide; Bortezomib; Carfilzomib
Introduction
Multiple myeloma (MM) is a hematological malignancy of plasma cells (PCs), characterized by lytic bone lesions, anemia, renal insufficiency, and the presence of monoclonal proteins in the blood and urine [1]. It is the second most common hematopoietic malignancy, with approximately 22,350 new cases and 10,710 deaths reported in 2013 [2]. The median age at diagnosis is approximately 70 years; 37% of patients are younger than 65 years, 26% are between the ages of 65 and 74 years, and 37% are 75 years of age or older [1]. Therapeutic advancement in recent years has increased the overall survival of myeloma patients. This is due not only to autologous Stem Cell Transplantation (SCT), but also to the availability of novel agents, such as the Immunomodulatory Drugs (IMiDs) thalidomide, lenalidomide, and pomalidomide, and the proteasome inhibitors bortezomib and carfilzomib [3]. Despite the use of these new agents, MM relapses in virtually all patients [4].
The unique feature of myeloma cells is their intimate relationship with the bone marrow (BM) microenvironment. This consists of various Extracellular Matrix (ECM) proteins, cell components, osteoclasts, and osteoblasts [5]. The BM microenvironment provides a specialized niche that supports growth of myeloma and maintain their long-term survival by secreting growth factors such as Interleukin-6 (IL-6), Insulin Like Growth Factor-1 (IGF-1), Vascular Endothelial Growth Factor (VEGF), B-cell Activating Factor (BAFF), Fibroblast Growth Factor (FGF), Stromal cell-Derived Factor 1a (SDF1a), and Tumor Necrosis Factor-a (TNFa) [6-10]. The direct interaction of the BM microenvironment with myeloma is responsible for myeloma cells growth and survival, angiogenesis, osteolytic lesions, and drug resistance [11-13]. Not surprisingly, multiple efforts have been made to target the interaction of myeloma cells to BM microenvironment for its treatment [14].
As in other malignant neoplasms, myeloma patients initially respond to chemotherapy; however, during the course of the disease, either intrinsic or acquired resistance to chemotherapy will emerge. MM is characterized by a complex genomic instability and cytogenetic constitution, which predisposes the malignant cells to intrinsic resistance [15-17]. The acquired resistance is mediated by several mechanisms, including overexpressed drug transporter proteins, such as P-glycoprotein (P-gp) -also known as Multidrug Resistance Protein 1 (MDR1)-[18], overexpression of anti-apoptotic proteins, such as B-cell leukemia protein 2 (Bcl-2), B-cell lymphoma-extralarge (Bcl-XL), Bcl2-related protein A1 (A1/Bfl1), and constitutive expression of transcription factor nuclear factor kappa B (NF-κB). Thus, therapeutic interventions that can induce apoptosis of tumor cells could become useful approaches to treat MM.
Intrinsic resistance in multiple myeloma
Genomic instability and complex cytogenetic constitution are hallmarks of MM. The genetic lesions leading to myeloma have been recognized as inherited variations, translocations, copy number abnormalities, mutations, methylation, and microRNA (miRNA) abnormalities [15,16,19]. These lesions are associated with the myeloma proliferation and drug resistance [15,17]. Based on karyotype abnormalities, MM can be divided in two main groups: hyperdiploid and non-hyperdiploid. Studies suggested that only 10% of hyperdiploid group show a primary IgH translocation at 14q32 locus, whereas 70% of cases in the non-hyperdiploid group carry that [20]. Of note, patients in the hyperdiploid group have a better survival and prognosis of those in the non-hyperdiploid group. The IgH translocation usually involves juxtaposition of the immunoglobulin gene to an oncogene on partner chromosomes creating several reciprocal translocations. The more frequent ones are the t(4;14) (p16;q32) and t(11;14)(q13;q32) translocations, present in 15% and 17% of myeloma patients, respectively [21]. There are several other translocations involving aberrant expression of oncogenes, such as c-MAF, c-Myc, and Cyclin D [21-24]. Whether these cytogenetic aberrations are directly associated with drug resistance and relapse is still debatable and warrants further investigations; nonetheless, recent studies have provided evidence that some of these cytogenetic abnormalities predispose to drug resistance and are associated to disease relapse. For example, studies of Chang-Yew et al. showed that blocking c-MAF, the oncogene overexpressed in patients with t(14;16) and t(14;20) translocations, inhibits cell proliferation and sensitizes myeloma cells to chemotherapy drugs [25,26]. Moreover, 1q21 gain, found in almost 70% of multiple myeloma patients harboring the t(14;16) or t(4:14) translocations, has been correlated with an adverse outcome [27,28].
Acquired resistance in multiple myeloma
Role of P-glycoprotein in acquired resistance
One of the most important players for acquiring drug resistance is the P-gp, produced by the MDR/ATP-Binding Cassette (ABC) B1 gene. This protein is the best known family member of pump transporters that mediate cellular efflux of peptides and drugs. Studies have shown that almost 75% of patients treated with doxorubicin, dexamethasone, and vincristine do increase the expression of P-gp [29]. Importantly, the expression of P-gp is found to be very low in untreated multiple myeloma patients, suggesting that the cumulative dose increases the expression of this protein in myeloma cells. A second drug-efflux protein, the Multidrug Resistance Protein-1 (MRP1), has not been found to be overexpressed in myeloma cells [30,31], making P-gp as the candidate protein in drug resistance. In untreated patients, P-gp does not seem to be expressed de novo. Interestingly, studies have shown that patients treated with melphalan do not increase P-gp expression [29], but increase in expression of P-gp was associated with treatment of other conventional chemotherapy agents, such as vincristine, doxorubicin, and dexamethasone [32], as well as novel agents, such as bortezomib and carfilzomib [33].
Because of the critical role of p-gp in drug resistance, inhibition of P-gp activity has become a major focus in clinical studies, and several MDR modulators have been tested to reverse drug resistance. For example, a phase I/II study was conducted using the MDR modulator verapamil, in combination with vincristine, doxorubicin, and dexamethasone, to treat patients with refractory MM [31]. This strategy induced a partial response rate of 50%, but this combination therapy resulted in excessive toxicity, and P-gp inhibitors do not constitute standard of care in the current management of MM.
Activation of nuclear factor-κB (NF-κB) and acquired resistance
Another major pathway that plays an important role in drug resistance is the NF-κB signaling pathway. The NF-κB term refers to a family of signal-responsive transcription factors that includes RelA/ p65, c-Rel, RelB, NF-κB1/p50 and NF-κB2/p52 [34]. In normal resting cells, NF-κB transcription factors are maintained in an inactive state within the cytoplasm through binding to inhibitory proteins called IκBa, which also include the unprocessed p105 and p100 forms of NF-κB1 and NF-κB2 [34]. The activation of NF-κB is divided into two pathways, canonical and non-canonical. The canonical pathway is induced by physiological NF-κB stimuli including TNF-a or IL- 1, and antigen receptors [35-37]. These cytokines bind and induce Tumor Necrosis Factor Receptor 1 (TNFR1) signaling. Stimulation of TNFR1 leads to the binding of tumor necrosis factor receptor Type1-Associated Death Domain protein (TRADD), which provides an assembly platform for the recruitment of Fas-Associated protein with Death Domain (FADD) and TNF Receptor-Associated Factor 2 (TRAF2) [38]. TRAF2 couples with receptor-interacting serine/ threonine-protein kinase 1 (RIP1) for IκB kinase (IKK) activation. IKK complex consists of two catalytically active kinases, IKKa and IKKβ, and the regulatory subunit IKKγ (NEMO) [39,40]. In the canonical pathway, IκBa is phosphorylated in an IKKβ- and NEMO-dependent manner followed by ubiquitination and proteasomal degradation, which thus releases the bound NF-κB dimers so they can translocate to the nucleus. In contrast, the non-canonical pathway, inducedby certain TNF family cytokines -such as CD40L, B-cell Activating Factor (BAFF) and lymphotoxin-β (LT-β)-, involves IKKa-mediated phosphorylation of p100 associated with RelB, which leads to partial processing of p100 and the generation of transcriptionally active p52-RelB complexes. IKKa activation and phosphorylation of p100 depends on Nuclear factor κB-Inducing Kinase (NIK), which is tightly regulated by TRAF3, TRAF2 and additional ubiquitin ligases [35-38]. Once in the nucleus, NF-κB dimers are further regulated by protein phosphorylation [41] and other post-translational modifications, such as protein acetylation [42], and activate genes whose products inhibit cell death [43-46], stimulate cell proliferation [47], and promote migratory and invasive phenotypes that are associated with tumor progression [48]. The transcription factor NF-κB could also contribute to drug resistance in myeloma through up-regulation of some Bcl-2 anti-apoptotic family members including Bcl-XL [49].
Myeloma cells often show constitutive expression of NF-κB [50]. Most solid and lymphoid tumors show constitutive NF-κB activity [51]. Constitutive or overexpression of NF-κB has been associated with myeloma cell proliferation, survival, invasion and drug resistance [52,53]. Although the precise role of NF-κB activation in pathogenesis of myeloma has not been fully characterized, it has been shown that myeloma cell adhesion to bone marrow stromal cells induces NF- κB-dependent up-regulation of transcription of IL-6 [54,55], which is a major growth and survival factor for myeloma cells. Since IL-6 upregulates adherence of myeloma cells to fibronectin and it confers resistance to drug-induced apoptosis, blockade of NF-κB signaling represents a novel therapeutic strategy in MM. Recent studies have indicated that targeting NF-κB pathway in myeloma has a positive outcome [56,57]. Along these line, myeloma cells harboring TRAF3 gene mutation in NF-κB pathway are resistant to dexamethasone but sensitive to bortezomib [58], suggesting that apoptotic function of bortezomib is partly explained by blocking the canonical NF-κB pathway; however, at the same time it unexpectedly induces the alternative (non-canonical) pathway, which makes myeloma cells less responsive [59]. Moreover, recent studies have shown that though canonical NF-κB pathway can be successfully blocked by small-molecule inhibitors of IKKß, in myeloma cell lines in vitro, the anti-MM activity in vivo of these IKKß inhibitors is limited because of the compensatory activation of the non-canonical pathway [60]. Furthermore, it has been reported that myeloma cells tend to develop a bortezomib-resistant NF-κB phenotype through a Proteasome- Inhibitor Resistant (PIR) pathway [61]. The latter flaws of bortezomib may explain to a large extent why it should be applied in combined regimens for those myeloma patients who are refractory to it. Since bortezomib-induced cytotoxicity cannot be fully attributed to inhibition of canonical NF-êB activity in myeloma cells, inhibition of both canonical and noncanonical pathways would be a desirable strategy to combat resistance.
Cancer Stem Cells and Resistance
The association of cancer stem cells (CSCs) and drug resistance has been demonstrated in various types of cancer including MM [62]. The importance of targeting CSC population has been realized for many years, but this research has been limited by the fact that a specific CSC population in MM has not been properly defined yet [17]. Studies from the last decade have supported the notion that CSCs are resistant to chemotherapeutic agents. Various strategies have been proposed to target the molecular, metabolic, and epigenetic signatures, and the self-renewal signaling pathway characteristic of CSCs. These cells exhibit high levels of ABC activity, Aldehyde Dehydrogenases (ALDH1), and Retinoic Acid (RA) Receptor a (RARa) that have been associated with clonogenic potential and resistance to chemotherapy [63,64]. Further research is needed to understand the molecular mechanisms that keep myeloma cells clonogenic and drive the transition between stem-like and non-stemlike states in the local bone marrow microenvironment. These studies could provide us new approaches to target drug resistance in MM.
Drug resistance to specific antineoplastic agents
Although we are far from having a satisfactory knowledge of the different mechanisms of resistance to various chemotherapy agents, multiple studies have elucidated particular strategies and mutations developed by myeloma cells after exposure to specific drugs. We herein describe the most important published experience with the various class of agents used in the treatment of MM. A few examples are shown in Figure 1.
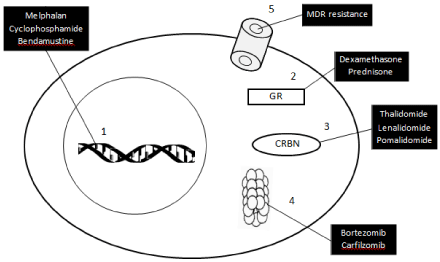
Figure 1: Examples of mechanisms of resistance of myeloma cells to
chemotherapeutics agents (details in text).
1: Increased repair of DNA interstrand cross links with alkylating agents;
2: Mutations of the hormone-binding region of the Glucocorticoid Receptor
(GR); 3: Decreased expression of the Cereblon (CRBN) protein with
the IMiDs drugs; 4: Mutations of the bortezomib-binding pocket of the
proteasome; 5: Multidrug Resistance (MDR), with cellular extrusion of a
variety of chemotherapy agents by the drug efflux transporter P-glycoprotein.
Glucocorticoids
Glucocorticoids, such as prednisone and dexamethasone, are among the most commonly used drugs in the treatment of MM. They are able to induce response rates of approximately 60% in newly diagnosed patients, even when used in monotherapy [65]. The Glucocorticoid Receptor (GR) is a member of the nuclear hormone receptor superfamily, which consists of structurally related proteins, that function as receptors for several hormones, including estrogens, progesterone, androgens, thyroid hormone, vitamin D, retinoids, and others. These cytoplasmic receptors are activated by the binding to their ligands, translocate to the nucleus, and regulate transcription of target genes that control biologic processes important in development, cell proliferation, and differentiation [66]. The nuclear hormone receptors typically consist of 5 domains: A/B (modulating region), C [DNA-binding region, containing two zinc-fingers which specifically recognize the Hormone-Responsive Element (HRE), a DNA enhancer sequence that is located near the promoter region of target genes], D (hinge region, important for nuclear localization), E (hormone-binding region, which also contains leucine zippers, the sites of dimerization), and F (modulating region). Following ligand binding, hormone-receptor complexes dimerize with other hormone-receptor complexes and translocate to the nucleus, where they bind to and initiate transcription of HRE expressing genes. The interaction between the receptor dimer and the HRE activates transcriptional machinery, leading to the expression of a specific set of genes. Resistance to corticosteroids may be due to mutations in their receptors: in a study of a myeloma line derived from a patient who became refractory to dexamethasone, the resistance was found to be caused by a truncated mRNA transcript of the GR, which lacked the C (hormone-binding) region [67]. Studies of Sanchez-Vega and Gandhi suggest that glucocorticoid resistance can also be mediated by transcription elongation block in the glucocorticoid receptor gene NR3C1 [68]. It has been proposed that glucocorticoid resistance should be divided in two forms, primary and secondary. Primary resistance is associated with the level of GR expression and the regulation of intracellular substrate availability [69]. The secondary form develops over the period of treatment, and its mechanism is still not completely understood. Clinical surveys suggest a correlation between functional GR expression levels and primary glucocorticoid sensitivity and prognosis [69].
Alkylating drugs
Alkylating drugs, such as melphalan and cyclophosphamide, are chemotherapy agents commonly used in the treatment of MM. Their mechanism of action involves the formation of cross links between the two strands of DNA (“inter-strand cross linking”), with impairment of DNA synthesis and cell replication. Melphalan, a drug synthesized in 1953, has a structure that incorporates two alkylating agents, nitrogen mustard and phenylalanine. More than 60 years later, this drug is still considered standard therapy for MM, either as single agent in the preparative regimen for autologous SCT, or in combination with other agents in SCT-ineligible patients [70]. Studies have indicated that the principal mechanism of resistance to melphalan is the increased repair rate of DNA inter-strand cross links [71,72], which is mediated by the Fanconi Anemia (FA)/BRCA pathway [73]. Silencing this pathway in melphalan-resistant cells with siRNA can reverse the drug resistance, whereas its overexpression promotes cell survival following melphalan treatment [73]. It is likely that the FA/BRCA pathway is involved even in cross-resistance to other alkylating agents and radiation therapy [74].
Proteasome inhibitors
The proteasome is a protein enzyme complex that breaks down and clears unused or misfolded proteins. Myeloma cells are particularly sensitive to inhibitors of the proteasome, presumably because they are specialized for the mass production of immunoglobulins. The increased protein load associated with this task lowers the threshold for proteotoxic stress and leaves plasma cells susceptible to toxic misfolded/unfolded proteins and pro-apoptotic signals initiated by the unfolded protein and endoplasmic reticulum stress responses [75]. Bortezomib is a dipeptide boronate that inhibits the ubiquitinmediated proteasome degradative pathway. The 26S proteasome consists of two 19S regulatory complexes and a barrel-shaped 20S proteolytic core. The 19S regulatory complexes bind the proteins tagged with ubiquitin, and direct them to the 20S core. The 20S is a proteolytic core that consists of 2a-subunit rings and 2ß-subunit rings, each of which contains 7 different a and ß subunits. The proteolysis is mediated by the ß-subunits: ß1 (caspase-like activity), ß2 (trypsinlike activity), and ß5 (chymotrypsin-like activity). Despite the efficacy of bortezomib in the treatment of myeloma, the neoplastic plasma cells invariably develop resistance to the boronic molecule. One study attributed the molecular resistance to bortezomib to a gene mutation (Ala49Thr substitution) of the proteasome ß5-subunit, which contains the bortezomib-binding pocket. The mutated protein was over expressed in resistant cells, and silencing of the ß5-subunit gene with siRNA restored bortezomib sensitivity and induced apoptosis [76]. This finding was confirmed by other studies, in which bortezomib resistance was caused by several other mutations involving the bortezomib-binding pocket of the ß5-subunit or its close proximity [77,78]. The development of novel proteasome inhibitors that bind to the a-subunits instead of the ß ones could overcome this mechanism of resistance.
Studies of bortezomib-resistant cell lines did not detect an abnormal composition or activity of the proteasome enzyme complex, and genomic profiling indicated the presence of other mechanisms of resistance, such as the overexpression of the Heat Shock Protein B8 (HSPB8), or cellular extrusion via the drug efflux transporter P-gp [79]. HSPB8 promotes the survival of myeloma cells by enhancing the autophagic removal of misfolded proteins through lysosomal degradation [80]. A second irreversible proteasome inhibitor, carfilzomib, has entered the clinical practice, and several other are currently in the development phase. Beside �5-subunit mutations and P-gp overexpression, mechanisms of resistance to these new agents have not yet been elucidated [33].
Immunomodulatory drugs (IMiDS)
Thalidomide and its derivatives lenalidomide and pomalidomide represent a new class of antineoplastic compounds called IMiDs, which have immune-modulatory, anti-inflammatory, and anti-angiogenic properties. Their mechanism of action was elucidated only recently, after the discovery of their receptor, the Cereblon protein (CRBN) [81]. CRBN is a substrate-recognition component of the E3 ubiquitin ligase complex, which includes the DNA Damage Binding protein-1 (DDB1), Cullin-4A (Cul4A), and Regulator of cullins (Roc1). It is now known that the therapeutic activity of IMiDs is due to a CRBN gain of function. IMiDs are able to modify the substrate specificity of CRBN, and induce the proteasome degradation of the ikaros proteins IKZF1 and IKZF3. These are two B cell transcription factors, which can be transcriptional activators or repressors, depending on different cellular settings [82,83]. In view of these discoveries, the IMiDs should be more properly called “ubiquitin ligase modulators”. In a study, resistance to lenalidomide was mediated by the induction of the Wnt/�-catenin pathway [84]. Other studies indicated that the resistance was mediated to decreased expression of the CRBN protein [85,86].
Conclusion
The introduction of novel agents in the treatment of MM, as the proteasome inhibitors and the IMiDs, has revolutionized its management, so that many patients are now a day able to remain in remission for several years. Results have been improved by the adoption of these novel agents, initially approved for the relapsing/ refractory setting, and later introduced into the upfront setting, and by incorporating them into combination regimens that produced unprecedented rates of disease response. Despite these therapeutic advances, disease relapse and progression occur almost universally, due to innate or acquired drug resistance.
In this review, we described several molecular mechanisms and pathways of resistance to different anti-neoplastic agents that have been elucidated so far. MM is a genetically complex cancer, characterized by intra-clonal heterogeneity and progressing with branching evolutionary patterns, instead of a linear multistep process [87]. According to this view, disappearance of sensitive neoplastic subclones after the initial chemotherapy can be followed by the emergence of other selected subclones with different mutations, and so on. While cells of an unmutated tissue respond homogeneously to a particular chemotherapy agent, the heterogeneity of multiple myeloma subclones may express different levels of drug sensitivity. At this stage, innate mechanisms mediated by MDR leads to crossresistance, and branching evolution with acquired mutations selects resistant clones that ultimately lead to the terminal phase of the disease. Resistance to a single drug can be eliminated by the use of combination regimens, using agents that target different receptors and molecular pathways inside the myeloma cell. Along these lines, specific cell signaling targeted therapies such as HDAC, PI3K/AKT/ mTOR, Hsp90, Wnt, Notch, Hedgehog; and strategies targeting the tumor microenvironment including hypoxia, angiogenesis, CD44, CXCR4, have yielded promising results alone or in combinations in preclinical or clinical studies involving patients with relapsed/ refractory MM. Based on these premises, we believe that a single general strategy to overcome drug resistance in all patients cannot be developed. However, with the rapid advances made by basic research and molecular oncology in the last decade, it is likely that the future of clinical management of myeloma patients will rely on the individualized application of genomic and proteomic analyses, targeting the vulnerable pathways of a specific patient in a specific phase of the disease. In order to cure MM, we need a better understanding of the complex drug resistance in this disease, andfurther development of new therapeutic agents is warranted.
References
- Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011; 364: 1046-1060.
- Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2013; 63: 11-30.
- Richardson PG, Mitsiades C, Schlossman R, Munshi N, Anderson K. New drugs for myeloma. Oncologist. 2007; 12: 664-689.
- Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008; 111: 2516-2520.
- Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007; 7: 585-598.
- Hideshima T, Chauhan D, Hayashi T, Podar K, Akiyama M, Gupta D, et al. The biological sequelae of stromal cell-derived factor-1alpha in multiple myeloma. Mol Cancer Ther. 2002; 1: 539-544.
- Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001; 98: 428-435.
- Vanderkerken K, Asosingh K, Braet F, Van Riet I, Van Camp B. Insulin-like growth factor-1 acts as a chemoattractant factor for 5T2 multiple myeloma cells. Blood. 1999; 93: 235-241.
- Freund GG1, Kulas DT, Mooney RA . Insulin and IGF-1 increase mitogenesis and glucose metabolism in the multiple myeloma cell line, RPMI 8226. J Immunol. 1993; 151: 1811-1820.
- Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006; 42: 1564-1573.
- Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004; 104: 607-618.
- Christian Jakob, Jan Sterz, Ivana Zavrski, Ulrike Heider, Lorenz Kleeberg, Claudia Fleissner, et al. Angiogenesis in multiple myeloma. Eur J Cancer. 2006; 42: 1581-1590.
- Roodman GD. Pathogenesis of myeloma bone disease. Blood Cells Mol Dis. 2004; 32: 290-292.
- Anderson KC. Oncogenomics to target myeloma in the bone marrow microenvironment. Clin Cancer Res. 2011; 17: 1225-1233.
- Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12: 335-348.
- Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood. 2009; 113: 2290-2297.
- Abdi J, Chen G, Chang H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget. 2013; 4: 2186-2207.
- Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993; 62: 385-427.
- Walker BA, Leone PE, Chiecchio L, Dickens NJ, Jenner MW, Boyd KD, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010; 116: 56-65.
- Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009; 23: 2210-2221.
- Boyd KD, Pawlyn C, Morgan GJ, Davies FE. Understanding the molecular biology of myeloma and its therapeutic implications. Expert Rev Hematol. 2012; 5: 603-617.
- Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R. Intergroupe Francophone du Myélome . Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood. 2001; 98: 3082-3086.
- Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A. 2000; 97: 228-233.
- Greco C, D'Agnano I, Vitelli G, Vona R, Marino M, Mottolese M, et al. c-MYC deregulation is involved in melphalan resistance of multiple myeloma: role of PDGF-BB. Int J Immunopathol Pharmacol. 2006; 19: 67-79.
- Chang-Yew Leow C1, Gerondakis S, Spencer A. MEK inhibitors as a chemotherapeutic intervention in multiple myeloma. Blood Cancer J. 2013; 3: e105.
- Popovic R, Licht JD. MEK and MAF in myeloma therapy. Blood. 2011; 117: 2300-2302.
- Chang H, Trieu Y, Qi X, Jiang NN, Xu W, Reece D. Impact of cytogenetics in patients with relapsed or refractory multiple myeloma treated with bortezomib: Adverse effect of 1q21 gains. Leuk Res. 2011; 35: 95-98.
- Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006; 108: 1724-1732.
- Grogan TM, Spier CM, Salmon SE, Matzner M, Rybski J, Weinstein RS, et al. P-glycoprotein expression in human plasma cell myeloma: correlation with prior chemotherapy. Blood. 1993; 81: 490-495.
- Linsenmeyer ME, Jefferson S, Wolf M, Matthews JP, Board PG, Woodcock DM. Levels of expression of the mdr1 gene and glutathione S-transferase genes 2 and 3 and response to chemotherapy in multiple myeloma. Br J Cancer. 1992; 65: 471-475.
- Yang HH, Ma MH, Vescio RA, Berenson JR. Overcoming drug resistance in multiple myeloma: the emergence of therapeutic approaches to induce apoptosis. J Clin Oncol. 2003; 21: 4239-4247.
- Cornelissen JJ, Sonneveld P, Schoester M, Raaijmakers HG, Nieuwenhuis HK, Dekker AW, et al. MDR-1 expression and response to vincristine, doxorubicin, and dexamethasone chemotherapy in multiple myeloma refractory to alkylating agents. J Clin Oncol. 1994; 12: 115-119.
- Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, et al. Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno) proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther. 2012; 341: 174-182.
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002; 109: 81-96.
- Bezbradica JS, Medzhitov R. Integration of cytokine and heterologous receptor signaling pathways. Nat Immunol. 2009; 10: 333-339.
- Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009; 228: 225-240.
- Kruglov AA, Kuchmiy A, Grivennikov SI, Tumanov AV, Kuprash DV, Nedospasov SA. Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: mouse models. Cytokine Growth Factor Rev. 2008; 19: 231-244.
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF- κB signaling pathways. Nat Immunol. 2011; 12: 695-708.
- Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008; 18: 334-342.
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997; 388: 548-554.
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009; 27: 693-733.
- Chen Lf, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001; 293: 1653-1657.
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996; 274: 782-784.
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996; 87: 565-576.
- Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996; 274: 787-789.
- Wang CY, Mayo MW, Baldwin AS. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996; 274: 784-787.
- Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 2001; 12: 73-90.
- Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004; 6: 297-305.
- Xiang Y, Remily-Wood ER, Oliveira V, Yarde D, He L, Cheng JQ, et al. Monitoring a nuclear factor-kappaB signature of drug resistance in multiple myeloma. Mol Cell Proteomics. 2011; 10: M110 005520.
- Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, et al. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004; 103: 3175-3184.
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002; 3: 221-227.
- Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol. 2011; 6: 249-274.
- Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood. 2010; 115: 3541-3552.
- Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, et al. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996; 87: 1104-1112.
- Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, et al. NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002; 277: 16639-16647.
- Lichtenstein A, Tu Y, Fady C, Vescio R, Berenson J. Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell Immunol. 1995; 162: 248-255.
- Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, Kroemer G. Targeting NF-kappaB in hematologic malignancies. Cell Death Differ. 2006; 13: 748-758.
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007; 12: 131-144.
- Fabre C, Mimura N, Bobb K, Kong SY, Gorgun G, Cirstea D, et al. Dual inhibition of canonical and noncanonical NF- κB pathways demonstrates significant antitumor activities in multiple myeloma. Clin Cancer Res. 2012; 18: 4669-4681.
- Hideshima T, Chauhan D, Kiziltepe T, Ikeda H, Okawa Y, Podar K, et al. Biologic sequelae of I{kappa}B kinase (IKK) inhibition in multiple myeloma: therapeutic implications. Blood. 2009; 113: 5228-5236.
- Markovina S, Callander NS, O'Connor SL, Kim J, Werndli JE, Raschko M, et al. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res. 2008; 6: 1356-1364.
- Abe M, Harada T, Matsumoto T. Concise review: Defining and targeting myeloma stem cell-like cells. Stem Cells. 2014; 32: 1067-1073.
- Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, et al. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008; 68: 190-197.
- Yang Y, Shi J, Tolomelli G, Xu H, Xia J, Wang H, et al. RARa2 expression confers myeloma stem cell features. Blood. 2013; 122: 1437-1447.
- Kumar S, Lacy MQ, Dispenzieri A, Rajkumar SV, Fonseca R, Geyer S, et al. Single agent dexamethasone for pre-stem cell transplant induction therapy for multiple myeloma. Bone Marrow Transplant. 2004; 34: 485-490.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell. 1995; 83: 835-839.
- Moalli PA, Pillay S, Weiner D, Leikin R, Rosen ST. A mechanism of resistance to glucocorticoids in multiple myeloma: transient expression of a truncated glucocorticoid receptor mRNA. Blood. 1992; 79: 213-222.
- Sánchez-Vega B, Gandhi V. Glucocorticoid resistance in a multiple myeloma cell line is regulated by a transcription elongation block in the glucocorticoid receptor gene (NR3C1). Br J Haematol. 2009; 144: 856-864.
- Shah DS, Kumar R. Steroid resistance in leukemia. World J Exp Med. 2013; 3: 21-25.
- Jagannath S, Kyle RA, Palumbo A, Siegel DS, Cunningham S, Berenson J. The current status and future of multiple myeloma in the clinic. Clin Lymphoma Myeloma Leuk. 2010; 10: 28-43.
- Spanswick VJ, Craddock C, Sekhar M, Mahendra P, Shankaranarayana P, Hughes RG, et al. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood. 2002; 100: 224-229.
- Laursen MB, Falgreen S, Bødker JS, Schmitz A, Kjeldsen MK, Sørensen S, et al. Human B-cell cancer cell lines as a preclinical model for studies of drug effect in diffuse large B-cell lymphoma and multiple myeloma. Experimental hematology. 2014; 42: 927-938.
- Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS . The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood. 2005; 106: 698-705.
- Gourzones-Dmitriev C, Kassambara A, Sahota S, Rème T, Moreaux J, Bourquard P, et al. DNA repair pathways in human multiple myeloma: role in oncogenesis and potential targets for treatment. Cell Cycle. 2013; 12: 2760-2773.
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006; 107: 4907-4916.
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008; 112: 2489-2499.
- Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, et al. Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012; 26: 757-768.
- Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A, et al. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia. 2010; 24: 1506-1512.
- Gutman D, Morales AA, Boise LH. Acquisition of a multidrug-resistant phenotype with a proteasome inhibitor in multiple myeloma. Leukemia. 2009; 23: 2181-2183.
- Hamouda MA, Belhacene N, Puissant A, Colosetti P, Robert G, Jacquel A, et al. The small heat shock protein B8 (HSPB8) confers resistance to bortezomib by promoting autophagic removal of misfolded proteins in multiple myeloma cells. Oncotarget. 2014; 5: 6252-6266.
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010; 327: 1345-1350.
- Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014; 343: 305-309.
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014; 343: 301-305.
- Bjorklund CC, Ma W, Wang ZQ, Davis RE, Kuhn DJ, Kornblau SM, et al. Evidence of a role for activation of Wnt/beta-catenin signaling in the resistance of plasma cells to lenalidomide. J Biol Chem. 2011; 286: 11009-11020.
- Zhu YX, Kortuem KM, Stewart AK. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk Lymphoma. 2013; 54: 683-687.
- Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012; 26: 2326-2335.
- Brioli A, Melchor L, Cavo M, Morgan GJ. The impact of intra-clonal heterogeneity on the treatment of multiple myeloma. Br J Haematol. 2014; 165: 441-454.