
Review Article
Ann Hematol Oncol.2015;2(5): 1041.
New Approaches to Predict Outcome and Personalize Therapy in Multiple Myeloma: from microRNAs to Integrated Genomics
Marco Rossi¹, Maria Teresa Di Martino¹, Pietro Hiram Guzzi², Pierosandro Tagliaferri¹ and Pierfrancesco Tassone¹,³*
¹Department of Experimental and Clinical Medicine, Magna Graecia University, Italy
²Department of Medical and Surgical Sciences, Magna Graecia University, Italy
³Sbarro Institute for Cancer Research and Molecular Medicine, Temple University, USA
*Corresponding author: Pierfrancesco Tassone, Department of Experimental and Clinical Medicine, Magna Graecia University, Viale Europa, 88100 Catanzaro, Italy
Received: May 05, 2015; Accepted: June 04, 2015; Published: June 06, 2015
Abstract
Multiple Myeloma (MM) is a plasma cell malignancy that arises from a preneoplastic condition known as monoclonal gammopathy of uncertain significance. Prognostic stratification relies on standardized staging system and the identification of specific chromosomal aberrations. The introduction of Gene Expression Profile (GEP) has provided new tools for outcome prediction. The recent discoveries on the role of micro RNAs in the pathogenesis of MM prompted the question whether they can be used to predict outcome and personalize therapy. In this work, we overview the current issues on the definition of clinical outcome in MM by miRNA profiling, and we discuss their potential role as anti-cancer therapeutics. Finally, we describe the integrative analysis that, by the means of conjugating the predictive power of GEP and miRNA profiles, will likely become the most relevant tool to warrant tailored therapy to MM patients in the next future.
Keywords: miRNAs; Non coding RNAs; Circulating miRNAs; Myeloma prognosis; Myeloma therapy; Integrative genomic analysis
Introduction
Current issues on diagnosis, prognostication and personalization of therapy in multiple myeloma
Multiple Myeloma (MM) accounts for about 10% of all hematological malignancies and is the natural evolution of a preneoplastic condition, known as Monoclonal Gammopathy of Uncertain Significance (MGUS). MGUS progresses to overt MM at a rate of 1% per year [1]. Symptomatic MM diagnosis relies on the presence of clonal Plasma Cells (PCs) >=10% of total nucleated bone marrow cells and end-organ damage signs including hypercalcemia, renal insufficiency, anaemia and bone lesions [2]. MM is associated with a heterogeneous pattern of chromosomal abnormalities that can be distinguished as complex Hyperdiploid (HD) kariotypes, that account for nearly 50% of cases or Non-Hyperdiploid (NHD) kariotypes [3]. HD abnormalities include typical translocations such as t(4;14), t(14;16), deletions of 13q, 17p and 1p and 1q chromosomal gains [4]. At the time of diagnosis, current recommendation for risk stratification includes International Staging System (ISS) definition, conventional karyotyping to identify trisomies, t(4;14) and chromosomes 13q /17p deletions, while FISH analysis should be performed for t(11;14), t(4;14), t(14;16), t(6;14), t(14;20), 17p del and 1q gains [2,5]. Although additional risk factors have been considered such as LDH levels, IgA isotype, and plasmablastic histology a general consensus on risk stratification is attributed to ISS and cytogenetics/ FISH. The m-SMART risk stratification developed at Mayo Clinic [6] integrates karyotype/FISH data to separate high [17p del; t(14;16);t(14;20)], intermediate [t(4;14); 1q gains; complex karyotype; 13q del or hypodiploidy] and standard risk patients [trisomies; t(11;14); t(6;14)]. As for ISS stage, high risk disease is usually observed for stages II and III.
These methods are widely used and routinely available for clinicians, but they cannot embrace the wide range of MM genetic abnormalities. Furthermore, novel biological agents, such as Immunomodulators (ImiDs) or Proteasome Inhibitors (PIs) require a deeper molecular analysis of MM-related genomic aberrations to provide better prognostication and prediction tools. In this light, Shaughnessy et al. [7] have analyzed the Gene Expression Profile (GEP) of 532 MM patients enrolled in 2 different trials. Long-term follow-up data were available from these studies, thus improving the quality of GEP analysis. Investigators used the log-rank test of expression quartiles to define a 70 gene-signature (GEP-70) that allowed identifying a patient population at high-risk of relapse and reduced Overall Survival (OS). Multivariate analysis that included poor prognosis translocations and ISS showed that GEP-70 was an independent predictor of clinical outcome. Interestingly, the GEP- 70 was able to better distinguish true low-risk patients, who have erroneously considered at high risk. Following clinical studies further validated GEP-70 in other MM cohorts [8,9]. GEP-70 has also shown power to predict outcome in MM patients treated with PIs or ImiDs [10-12]. However, these earlier investigations could not identify a clear cut distinction between MGUS and MM patients by GEP-70. A recent work prospectively evaluated MGUS patients from a large US cooperative trial and found that GEP-70 signature was an independent predictor of progression to symptomatic MM. A risk model based on GEP-70, serum free light chain levels and monoclonal component spike identified a group of patients at high-risk of progression to MM [13]. These findings support the use of GEP for risk stratification and prediction of clinical outcome for MM. A relevant concern is related to the difficulty to extend this method to common routine clinical practice. Indeed, current use of GEP is confined to clinical trials, even if a risk score model such as m-SMART adopts GEP to define the high-risk group [6].
Emerging data on non-coding RNAs in diagnostic/prediction phases and as potential therapeutic tools in MM increases the complexity of management of this disease. In the next sections, we provide an overview of the state-of-art of microRNAs (miRNAs) in MM, including their pathogenetic and clinical role. We propose the rationale and describe the methods for an integrated genomic approach in MM. We believe that this approach would better answer to the critical and debated issues of prognosis and best affordable tailored therapy for MM patients.
The role of miRNAs in the pathogenesis of MM
miRNAs are endogenous short non coding RNAs (~22 nt) that control gene expression at the post-transcriptional level by targeting the 3’-UTR complementary sequence of mRNAs leading to inhibition of protein translation. miRNA sequences represent approximately 1% of the genome of different species. Mature miRNAs arise from a two step process, where the primary 70-100 nt miRNAs (pri-miRNAs), transcribed from genes located within extra or intragenomic regions, are detected and cleaved in the nucleus by the ribonuclease DROSHA. The resulting pre-miRNA precursors undergo a second cleavage in the cytoplasm by the RNA Pol III DICER into 18-24 nucleotidelong miRNA duplexes. Only one of the 2 strands (guide strand) is driven by the miRNA-containing RNA-Induced Silencing Complex (miRISC) to the 3’-UTR mRNA target sequence. The other strand, indicated as miRNA*, is degraded. The guide strand-mRNA binding inhibits the translation or promotes the decay of the targeted mRNA. Based on this mechanism, a single miRNA can control different target genes [14-18]. miRNA expression pattern is deeply deregulated in MM. Several studies have analyzed the global miRNA expression in malignant PCs (MM cell lines and/or primary PCs) compared with MGUS and healthy subjects [19-22]. These data indicate that MM PCs-derived miRNAs are preferentially up regulated rather than down regulated. Furthermore, higher total miRNA expression significantly correlated with the expression of genes involved in cancer initiation and progression [22]. miRNA expression seems to be related also to genomic aberrations. Indeed, Lionetti et al. [21] distinguished 48 MM and 6 PC leukemia patients based on Translocation/Cyclin (TC) classification. Twenty-six miRNAs were selected as differentially expressed across the 5 TC groups, demonstrating that defined chromosomal abnormalities may be associated with specific miRNA deregulation rather than only gene expression.
The pattern of dysregulated miRNAs strongly contributes to MM development and progression. The emerging picture from these studies shows that up regulated miRNAs support cancer growth and progression (onco-miRNAs), while down regulated ones should work as classical tumor suppressors (TS-miRNAs). This hypothesis represents the base to use miRNAs as prognostic/predictive and therapeutic tools. The next section will explore the most recent studies that focus on these topics.
miRNAs as prognostic tools: the promises and challenges of circulating miRNAs
Cancer biomarkers are valuable tools in the diagnostic, therapeutic and follow-up phases of the disease. A biomarker can be used alone or in combination to predict the patients prognosis. To accomplish these goals, the biomarker has to be highly sensitive, specific, while its source must be easily accessed without harming the patient. For these reasons, serum markers have been often preferred for cancer patients. Starting from early findings showing miRNA deregulation as a hallmark of cancer [23,24], investigators evaluated whether miRNAs could turn to be potential biomarkers. In 2008, different groups separately identified the presence of circulating miRNAs in sera/plasma of cancer patients [25,26,27]. These studies showed that circulating miRNAs are stable molecules that cannot be degraded by RNAses. This is relevant as RNAses are highly abundant in the plasma [28]. The intrinsic resistance to degradation of serum/plasma miRNAs is due to the fact that these molecules circulate into body fluids through lipid based carriers and lipid free proteins [29,30], such as exosomes, HDL, LDL and AGO2 proteins. Selection of circulating miRNA carriers allows to discriminate true cell free miRNAs from blood cells derived ones. For instance, exosomes can be separated from whole blood by ultracentrifugation and antibody targeting [31], while protein carried-miRNAs are isolated by antibody-based precipitation of the chaperon protein. Therefore, cell free miRNAs are the most suitable candidate as cancer biomarkers [30]. In the great variety of tumors, serum/plasma miRNAs cannot be easily compared to cancer-derived miRNAs isolated from single patients. However, MM represents an important exception as malignant PCs can be collected from bone marrow and miRNA content can be evaluated. Interestingly, several studies have shown that miRNA profiles in the circulation and within malignant PCs are not overlapping [32,33]. These data suggest that serum/plasma miRNA deregulation is promoted by MM, but the resulting miRNA pattern is not exclusively due to the release of PCs-derived miRNAs into the body fluids. Circulating miRNAs can discriminate MM/MGUS and healthy donors as their profile are quite divergent [33]. However, there is no clear-cut separation between MM and MGUS samples, indicating that circulating miRNA profile of the premalignant condition MGUS will be associated also to MM progression [33]. When considering the specific circulating miRNA profile isolated from MM/MGUS patients, the studies appear to disclose divergent scenarios. This is likely due to different methods followed to attain the patient miRNome. Jones et al. [32] distinguished 4 preliminary groups to be analysed: healthy donors, non MGUS/non MM patients, MM with low and high paraprotein levels (<10 g/L or > 20 g/L, respectively). miRNAs were directly extracted from sera and a microarray analysis was performed to evaluate the miRNA profile in these groups. Only 9 miRNAs were chosen as consistently represented in all groups. These miRNAs underwent further validation through quantitative RT-PCR. Six out of 9 miRNAs were confirmed by RT-PCR. From this pool, investigators chose miR-720, miR-1246 and miR-1308 for further evaluation as they were expressed at high levels and with significantly different pattern between patients and controls. The authors also claimed that in a subsequent evaluation, miR-1308 was found to be not a miRNA, but a 5’-cleaved fragment of a tRNA, but they anyway included it in the analyses. To perform a correct comparison, the absolute concentration of each miRNA was determined (i.e. the copy number per microliter for each miRNA). This is a relevant point as standard normalisers for serum miRNAs are not currently available. Indeed, the usual standards for quantitative PCR such as RNU44 are not present in the serum [34]. Interestingly, miR-720 and miR-1308 levels discriminate between MM/MGUS and healthy groups, either alone and in combination. Combination of miR-720 and miR-1246 is also able to separate MM/MGUS from controls, while miR-1246 alone does not retain such power. In this study, the tested miRNAs did not correlate with paraprotein levels, except for miR-1246 (slight significance or negative correlation) [32]. A larger cohort of patients was analyzed to identify circulating miRNAs with a prognostic value in a subsequent and recently published work [33]. A hundred-three newly diagnosed MM and 18 relapsed MM patients were enrolled in the study together with 57 MGUS and 30 healthy donors. Circulating miRNAs were extracted from serum; for 6 newly diagnosed MM patients, miRNAs from exosomal and not exosomal fraction and bone marrow PCs were also obtained. A large number of miRNAs (667) were screened by Taq Low Density Array on representative samples for each condition. Seven miRNAs (miR-222, miR-744, miR- 34a, miR-130a, let-7d and let-7e) were selected according to different expression levels between MM/MGUS and control groups and then validated by qRT-PCR. The levels of each of these selected miRNAs were able to discriminate MM/MGUS patients from controls. As mentioned above, none of the selected miRNAs separated MM from MGUS patients. The 7 miRNAs correlated with ISS parameters and with plasma creatinine. Only let-7e showed a correlation with Durie-Salmon staging system. No correlation with percentage of PC infiltration was found, while low levels of let-7e were associated with a specific chromosomal aberration, the del (13q14). When considering the expression of these miRNAs within exosomal and non-exosomal fractions, the selected miRNAs were preferentially enriched in the exosomal fraction with the exception of miR-34a. The exosomal fraction retained lower levels of these miRNAs as compared to PCs. No correlation was found between exosomal and PC miRNAs, indicating that the release of circulating miRNAs cannot be dependent only on miRNA content of malignant PCs. However, the most relevant finding of this study is the correlation between selected miRNAs and patient outcome. Indeed, reduced levels of miR-744 and let7e were associated with worse OS and TTP. The authors underline that miR-744 maps within 17p12 region, which is close to TP53 locus (17p13), although a clear cut relationship between low miR-744 and TP53 deletion could not be proven. Furthermore, low levels of miR- 744 were associated with chromosomal 1q21 amplification or t(4;14) [35,36]. Overall, these data strongly support the use of circulating miRNAs as biomarkers to predict MM outcome and to discriminate healthy from MM/MGUS patients. For these purposes, exosomal miRNA fraction seems to be the preferred source of circulating miRNAs. On the other hand, circulating miRNAs selected in these studies cannot clearly discriminate MGUS from MM patients and do not exactly resemble miRNA content in malignant PCs.
Potentials and limits of miRNAs-based therapy
In 2009, Slack and Duchaine [37], proposed to use miRNAs as therapeutic tools for cancer. Their hypothesis was based on the notion of miRNA deregulation in human cancers and the possibility to identify significantly up- or downregulated miRNAs (onco-miRNAs and TS-miRNAs, respectively) within tumor cells as compared to normal counterpart. On this basis, miRNA inhibitors could be adopted to inhibit upregulated oncomiRNAs or miRNA mimics may replace downregulated TS-miRNAs [38]. This simple and logical approach has been recently investigated and relevant data have been produced since then. Early findings by Pichiorri et al. [19,20] showed that deregulated miRNAs of malignant PCs could be targeted to restore p53 mediated anti proliferative activity. MiR-15 and -16 have been described as TS-miRs in CLL [39], and were evaluated in MM setting. MiR-15 and -16 replacement was able to inhibit MM cell proliferation in vitro [40]. Both miRNAs target VEGF in MM cells interfering with neoangiogenesis [41]. Interestingly, loss of miR-15 in bone marrow stromal cells derived exosomes favoured the proliferation of MM cells [42] and miR-15 and -16 replacements was a valuable anti-MM strategy. Along with miRNAs-15 and -16, miR-29b seems a suitable candidate for miRNA-based replacement therapy. Indeed, miR-29b has shown anti-proliferative and proapoptotic activity on MM cells [43,44]. More importantly, miR-29b targets epigenetic regulators such as DMTAs that are involved in MM development and progression [45]. miR-29b replacement potentiates bortezomib activity and interferes with MM cell migration [46]. In the context of MM microenvironment, we have demonstrated that miR-29b reduces Osteoclastic (OCL) mediated bone resorption by targeting c-FOS within OCLs [47]. Potent anti-MM effects were observed in the presence of increased levels of miR-34a both in vitro and in vivo in a SCID model and in a SCID-synth-hu model, where human MM cells proliferate within a bio-polimeric support [48- 52]. Raimondi et al. have also demonstrated that neoangiogenesis can be efficiently targeted by miR-based therapies with intriguing results both in vitro and in vivo [53]. Along with miRNA replacement therapy, our group sought to investigate the efficacy of miRNA inhibitory molecules (antagomiR approach). In this light, inhibition of miR-21 [54] has shown promising results, as miR-21 support of MM cell growth is also dependent on BMSCs. Finally, miR-221/222 antagonism demonstrated a remarkable anti MM activity especially in poor prognostic MM group, carrying the t(4;14) [55,56]. Recently, an antagomir approach has also been used to restore p53 activity and exert antiproliferative effects on MM cells [57]. These data have confirmed that a miRNA-based therapy is feasible and effective in MM setting, but they have also raised important issues that represent the next future challenge in developing miRNA therapeutics. Indeed, the pleiotropic effects of miRNAs due to multiple targeting have shown that a real prediction of molecular consequences of miRNA replacement or antagonism is difficult to determine. We and others have described anti-MM effects, demonstrating the interference with relevant intracellular target proteins. Such an approach, although methodologically correct, does not allow to unveil the complex connections established by miRNA modulation within the tumor cells. This point is crucial as Molecularly Targeted Drugs (MTDs) have shown important limitations and in most cases did not meet the expectations in the clinical setting [58]. As an example, we have shown that targeting MM cells with a MTD such as valproic acid determines an important modulation of GEP in vivo, magnifying the number of pathways, that were likely to be involved, when considering the mechanism of action of the drug [59]. Therefore, these findings indicate that miRNA therapies produce anti-tumoral effects that are even beyond the Slack paradigm [37] and require to be evaluated with a more complex approach. On this regard, the possibility to integrate the information deriving from GEP and miRNA profiling provides investigators a new and powerful tool to develop ad hoc anti-cancer therapies.
Personalization of therapy: The era of integrated genomic approaches in cancer
Currently, a variety of technological platforms have enabled researchers to collect extensive information about different aspects of molecular biology. The rising body of data has led to the emergence of the computational integrative genomics, or integromics, a novel discipline in which computer science, bioinformatics and mathematical modeling, play a synergistic role in the interpretation of large datasets belonging to different data sources [60,61]. The focus of computational integrative genomics is to discover basic principles of interplay of different molecules in order to better elucidate molecular mechanims under the assumption that the information gathered from integrated analysis is higher than in the separate study of any data source [62]. Data sources of integromics are related mRNA, miRNA and protein expression, DNA copy number, SNPs, and may be produced in dedicated experiments or extracted by different databases. The scientific community has recently produced a large number of different databases that could be used in theory for integrated analysis. In addition to academic data, pharmaceutical and biotech companies retain large amounts of �proprietary data’ - inherited from their own and other sources. Most of data is stored in older types of databases designed to manage a single type of data, therefore the integration of these data source into a single comprehensive one is a relevant challenge [63]. The exhaustive enumeration of all these approaches is beyond the scopes of this review. Here, we summarize some recent approaches that integrate different data source, useful for researchers that we could categorize on following groups: (i) Big projects aiming to provide comprehensive resources for cancer studies (e.g. The Cancer Genome Atlas [TCGA], or the International Cancer Genome Consortium ICGC [ICGC]) and a set of smaller projects focusing on some narrow aspects of cancer research. For the purposes of this review, we will focus on the second class. Main results of these efforts are: (i) the individuation of possible biomarkers or synergistic regulatory mechanisms among molecules, (ii) the building of software tools able to perform joint analysis from different data sources as discussed later. Camps et al. [64], provided an integrated analysis of miRNA and mRNA expression and association with Hypoxia Inducible Factor (HIF) binding to study miRNA expression under hypoxia. Authors use MCF-7 breast cancer cells under hypoxic and normal oxygen conditions at three different time points with the following technologies: siRNA against HIF-1a and HIF-2a, miRNA expression through microarray technology, small RNA sequencing, gene expression profiles by microarrays and real-time polymerase chain reaction (rt-PCR). Moreover, previous published datasets have been used to build a model to correlate changes on miRNA and mRNA expression associated to HIF binding. This analysis led the authors to conclude that integrated analysis of microRNA, mRNA and ChIP-seq data into a single model supports the hypothesis that miRNA expression under hypoxic conditions is regulated at transcriptional and post-transcriptional levels. Kim et al. [65], focused on cancer survival aiming to increase the predictive accuracy of survival classes by integrating miRNA and mRNA data. They developed a novel machine learning approach based on feature selection with Cox proportional hazard regression model (FSCOX) to improve the prediction of cancer survival time. They compared the ability of this model to build survival curves using 3 types of cancer tissue data sets: (i) miRNA expression, (ii) mRNA expression, and (iii) combined miRNA and mRNA expression. Results evidenced that the integrated dataset yielded better results than the individual data sets, suggesting that interactions between miRNA and mRNA features, that are not detectable in the individual analyses, play an important role in this biological system.
In parallel to the so far discussed approaches, a remarkable number of software tools available to researchers have been produced. Main elements of these tools are:
- the ability to receive as input different data-sources: (e.g.) miRNA expression levels obtained using Microarray Technologies or Next Generation Sequencing (NGS); mRNA expression levels; copy number data;
- a robust statistical model able to gather information from different data sources;
- a knowledge base containing known associations among molecules (e.g. Transcription Factors);
- the ability to give semantics to results through ontologies;
- the implementation as available tool.
Such elements are generally integrated in a comprehensive pipeline whose general schema is reported in the Figure 1.
When considering specific software tools able to integrate in a single model miRNA and mRNA data (Table 1), we should recall: the approach of Gade et al, dchipGemiNI, MAGIA2 and mirCONNx. These approaches are based on the rationale exposed on Figure 1 and differ in general in the statistical/computational model used to merge data.
![]()
Tool
Input
Output
WebSite
Model
dCHIPGemini
miRNA/mRNA Expression Data Time Series
Feed Forward Loops FFL
Statistical Model and Literature Evidence
MAGIA2
miRNA/mRNA Expression Data Time Series
Feed Forward Loops FFL
Ontological AnalysisStatistical Model and Literature Evidence
mirConnX
miRNA,mRNA time series
Regulatory Networks
Pre Built Network.
miRIN
miRNA,mRNA
Regulatory networks of miRNA, mRNA, TFs and proteins.
Associations derived from literature
Table 1: Available software tools that integrate in a single model miRNA and mRNA data.
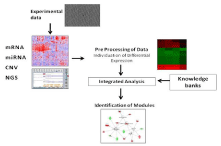
Figure 1: This picture depicts the flow of data in Integromics. Different
experimental data are collected from the investigator. The data span from
classical microarray technologies (e.g. mRNA or miRNA micro arrays) to next
generation sequencing techniques as well as genomic technologies such as
CNV or SNP arrays. The whole set of data is then preprocessed in order
to select only significant subsets of data or to evidence difference among
classes. Then data are integrated into single theoretical models and analyzed
with respect to data and information contained in existing knowledge
repositories. Finally, results are presented to the users by using supporting
models usually coming from graph theories.
Gade et al. [66] proposed a method to fuse miRNA and mRNA data into one prediction model using graph theory. Authors demonstrated that the integrated analysis may improve the clinical outcome providing better accuracy in survival prediction.
The dchipGEMINI [67] is a freely available web server that receives as input expression levels of miRNA and mRNA obtained from time series experiments analysing two conditions, e.g. normal and cancer conditions. It is able to individuate Feed-Forward Loops (FFLs) consisting of Transcription Factors (TFs), miRNAs and their common target genes. The association among miRNA and their target (TF and mRNA) information’s are obtained from the literature and stored into the web server. TFs derived from literature as used as null model to statistical ranks predicted FFLs from the experimental data.
Magia2 [68] represents the evolution of the precedent MAGIA web tool for the integrated analysis of both genes and microRNA. MAGIA receives as input miRNA and mRNA expression levels obtained by time-series experiments. It is able to associate miRNA and mRNA levels by integrating literature evidence, prediction algorithms, and experimental data through miRNA-target expression anti-correlation using four different relatedness measures. It is able to highlight different regulatory circuits involving either miRNA or TF as regulators: i) a Transcription Factor (TF) that regulates both a given miRNA and its target gene; ii) a miRNA that regulates both a given TF and its regulated gene. Furthermore, this tool provides functional enrichment of the gene network using DAVID platform.
mirConnX [69] is a software tool based on a web interface to build gene regulatory networks starting from mRNA and microRNA expression data on a whole genome scale. It is based on a built network used as a priori model consisting of Transcription Factor (TF)-gene associations and miRNA target predictions for human and mouse derived by computational methods and literature. Experimental data are used to infer experimental associations among TF, miRNA and genes. Associations are then used to weight the pre-defined network and resulting weighted network is visualized to the user.
miRIN is a web application designed for the identification of the modules of protein-protein interaction networks regulated by miRNAs. The approach of analysis consists of the integration of miRNA target data from literature, protein�protein interactions between target genes from literature, as well as mRNA and miRNA expression profiles provided as input. The output of miRIN is a set of regulatory networks involving miRNAs, mRNAs, TFs and proteins.
Integrated genomics in MM
In this section, we briefly recall some recent approaches of integrated analysis that focused mainly on MM. As general consideration, we should note that each of these has a tailored pipeline of analysis slightly different from what we have exposed so far. Approaches differ based on the aims of the work and consequently on input data and on analysis methodology. Considering the temporal evolution of the approaches, we may recall that first approaches of integration of data aimed to determine association among miRNA and mRNA by using experimental data of expression level to correlate changes of miRNAs levels with changes in mRNA levels. Following approaches aimed to integrate in a single model more than two data sources are intended to highlight more complex regulatory approaches. For instance, Huang et al. [70] used for the first time both mRNAs and miRNAs data. They focused on the identification of functional miRNA-target relationships excluding TF or protein data. Similarly, Ruike et al. [71] used miRNA and mRNA data obtained from K562 cell lines to predict functional association among molecules. Gutierrez et al. [72] used miRNA and mRNA expression levels of 60 MM patients to determine the relationships among types and changes in miRNA levels and the different cytogenetic subtypes, thus identifying disease related miRNAs and genes. They integrated target prediction and anti-correlation using the Pearson correlation coefficient to identify the over-represented pathways [21,72,73]. The Pearson correlation coefficient represents the closeness of related miRNA-target pair. As multiple miRNAs can target the same gene, the regulation effect can be cumulative. However, the existing pathway identification methods have omitted the cumulative effect, and considered neither the alteration of target gene’s expression nor the simultaneous cumulative regulatory effects. However, on these premises, Lionetti, et al. [21] performed an integrated analysis, based on computational target prediction and miRNA/mRNA profiling, and built up a network of predicted functional miRNA-target regulatory relations evidenced by expression data. In addition, the integrative analysis including genome-wide copy number profiles, provided new insights in the molecular MM subtypes. Recently, Zhang et al. [74] used MM data as a case study of an integrative approach of analysis aiming to discover different miRNA-mRNA regulatory mechanisms in different subtypes of samples. The authors proposed a genetic algorithm to identify specific miRNA-mRNA Functional Regulatory Modules (MFRMs) associated with different subtypes of this heterogeneous disease. The integrative analysis was performed by integration of three biological data sets: GO biological processes, miRNA target information, and matched miRNA and mRNA expression data. The proposed model was able to highlight the active miRNAs/mRNAs pairs and the mechanism that leads to specific differences for each subtype. By this approach, it is possible to identify the few miRNAs and mRNAs that act as “hub” of the biological processes involved in MM development and progression, because it included most of the MFRMs that can be potential biomarkers to discriminate MM subtypes. Finally, sample-matched miRNA (miRNAs)-mRNA expression data of multiple myeloma and prostate cancer have proven to be effective and reliable in identifying disease risk pathways that are regulated by miRNAs [15].
Conclusion
Understanding the functions of miRNAs in MM pathogenesis is extending our comprehension of tumor development and progression at a terrific pace. The dysregulation of miRNA expression is correlated with genomic aberrations and the intracellular pathways that strongly support malignant PC growth. Furthermore, miRNA profiles in the body fluids, although not completely concordant with malignant PC miRNA content, are modulated before and after therapy. These findings support their use as innovative biomarkers and potential therapeutic tools. More importantly, miRNA profiles can be integrated in a complex network of high-throughput arrays that include GEP and proteomic assays, to reveal the different molecular profiles and distinguish several subtypes of the disease contributing to better describe the biological behaviour of malignant PCs and to translate molecular findings into clinical useful advances. From this point of view, integrative analysis of the genetic events and the regulation of gene expression including epigenetic modifications, takes into account the structural information of pathways and the regulatory networks simultaneously. Integration of platforms and data (integromics approach) could be considered the key element to go deeply in the biology of the disease. The range of information derived from integromics approach would help to design the best affordable tailored therapy with the lowest toxicity profile to treat MM patients.
Acknowledgement
This work has been supported by the Italian Association for Cancer Research (AIRC), PI: PT. “Special Program Molecular Clinical Oncology - 5 per mille” n. 9980, 2010/15.
References
- Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002; 346: 564-569.
- Vincent Rajkumar S. Multiple myeloma: 2014 Update on diagnosis, risk-stratification, and management. Am J Hematol. 2014; 89: 999-1009.
- Tassone P, Tagliaferri P, Rossi M, Gaspari M, Terracciano R, Venuta S. Genetics and molecular profiling of multiple myeloma: novel tools for clinical management? Eur J Cancer. 2006; 42: 1530-1538.
- Morabito F, Recchia AG, Mazzone C, Gentile M. Targeted therapy of multiple myeloma: the changing paradigm at the beginning of the new millennium. Curr Cancer Drug Targets. 2012; 12: 743-756.
- Munshi NC, Anderson KC, Bergsagel PL, Shaughnessy J, Palumbo A, Durie B, et al. Consensus recommendations for risk stratification in multiple myeloma: report of the International Myeloma Workshop Consensus Panel 2. Blood. 2011; 117: 4696-4700.
- Mikhael JR, Dingli D, Roy V, Reeder CB, Buadi FK, Hayman SR, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clinic proceedings. 2013; 88: 360-376.
- Shaughnessy JD, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. Stewart JP, Kordsmeier B, Randolph C, A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007; 109: 2276-2284.
- Waheed S, Shaughnessy JD, van Rhee F, Alsayed Y, Nair B, Anaissie E, et al. International staging system and metaphase cytogenetic abnormalities in the era of gene expression profiling data in multiple myeloma treated with total therapy 2 and 3 protocols. Cancer. 2011; 117: 1001-1009.
- Nair B, Shaughnessy JD, Zhou Y, Astrid-Cartron M, Qu P, van Rhee F, et al. Gene expression profiling of plasma cells at myeloma relapse from tandem transplantation trial Total Therapy 2 predicts subsequent survival. Blood. 2009; 113: 6572-6575.
- Burington B, Barlogie B, Zhan F, Crowley J, Shaughnessy JD. Tumor cell gene expression changes following short-term in vivo exposure to single agent chemotherapeutics are related to survival in multiple myeloma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008; 14 : 4821-4829.
- Kumar SK, Uno H, Jacobus SJ, Van Wier SA, Ahmann GJ, Henderson KJ, et al. Impact of gene expression profiling-based risk stratification in patients with myeloma receiving initial therapy with lenalidomide and dexamethasone. Blood. 2011; 118: 4359-4362.
- Shaughnessy JD, Qu P, Usmani S, Heuck CJ, Zhang Q, Zhou Y, et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with Total Therapy 3. Blood. 2011; 118: 3512-3524.
- Dhodapkar MV, Sexton R, Waheed S, Usmani S, Papanikolaou X, Nair B, et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (SWOG S0120). Blood. 2014; 123: 78-85.
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116: 281-297.
- Ambros V. The functions of animal microRNAs. Nature. 2004; 431: 350-355.
- Amodio N, Di Martino MT, Neri A, Tagliaferri P, Tassone P. Non-coding RNA: a novel opportunity for the personalized treatment of multiple myeloma. Expert Opin Biol Ther. 2013; 13: S125-S137.
- Rossi M, Amodio N, Di Martino MT, Tagliaferri P, Tassone P, Cho WC. MicroRNA and Multiple Myeloma: from Laboratory Findings to Translational Therapeutic Approaches. Current pharmaceutical biotechnology. 2014; 15: 459-67.
- Rossi M, Amodio N, Di Martino MT, Caracciolo D, Tagliaferri P, Tassone P. From target therapy to miRNA therapeutics of human multiple myeloma: theoretical and technological issues in the evolving scenario. Current drug targets. 2013; 14: 1144-1149.
- Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell. 2010; 18: 367-381.
- Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105: 12885-12890.
- Lionetti M, Biasiolo M, Agnelli L, Todoerti K, Mosca L, Fabris S, Sales G, Deliliers GL, Bicciato S, Lombardi L, Bortoluzzi S, Neri A (2009) Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 114 (25):e20-26. doi:10.1182/blood-2009-08-237495
- Zhou Y, Chen L, Barlogie B, Stephens O, Wu X, Williams DR, Cartron MA, van Rhee F, Nair B, Waheed S, Pineda-Roman M, Alsayed Y, Anaissie E, Shaughnessy JD, Jr. (2010) High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proceedings of the National Academy of Sciences of the United States of America 107 (17):7904-7909. doi:10.1073/pnas.0908441107
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006; 6: 259-269.
- Calin GA, Croce CM MicroRNA. signatures in human cancers. Nat Rev Cancer. 2006; 6: 857-866.
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proceedings of the National Academy of Sciences of the United States of America 105 (30):10513-10518. doi:10.1073/pnas.0804549105
- Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, Banham AH, Pezzella F, Boultwood J, Wainscoat JS, Hatton CS, Harris AL (2008) Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. British journal of haematology 141 (5):672-675. doi:10.1111/j.1365-2141.2008.07077.x
- Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008; 18: 997-1006.
- Tsui NB, Ng EK, Lo YM. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin Chem. 2002; 48: 1647-1653.
- Vickers KC, Remaley AT. Lipid-based carriers of microRNAs and intercellular communication. Curr Opin Lipidol. 2012; 23: 91-97.
- Mo MH, Chen L, Fu Y, Wang W, Fu SW. Cell-free Circulating miRNA Biomarkers in Cancer. J Cancer. 2012; 3: 432-448.
- Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008; 110: 13-21.
- Jones CI, Zabolotskaya MV, King AJ, Stewart HJ, Horne GA, Chevassut TJ, et al. Identification of circulating microRNAs as diagnostic biomarkers for use in multiple myeloma. Br J Cancer. 2012; 107: 1987-1996.
- Kubiczkova L, Kryukov F, Slaby O, Dementyeva E, Jarkovsky J, Nekvindova J, Radova L, Greslikova H, Kuglik P, Vetesnikova E, Pour L, Adam Z, Sevcikova S, Hajek R (2014) Circulating serum microRNAs as novel diagnostic and prognostic biomarkers for multiple myeloma and monoclonal gammopathy of undetermined significance. Haematologica 99 (3):511-518. doi:10.3324/haematol.2013.093500
- Zhu W, Qin W, Atasoy U, Sauter ER. Circulating microRNAs in breast cancer and healthy subjects. BMC Res Notes. 2009; 2: 89.
- Nemec P, Zemanova Z, Greslikova H, Michalova K, Filkova H, Tajtlova J, Kralova D, Kupska R, Smetana J, Krejci M, Pour L, Zahradova L, Sandecka V, Adam Z, Buchler T, Spicka I, Gregora E, Kuglik P, Hajek R (2010) Gain of 1q21 is an unfavorable genetic prognostic factor for multiple myeloma patients treated with high-dose chemotherapy. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 16 (4):548-554. doi:10.1016/j.bbmt.2009.11.025
- Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, et al. R In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003; 101: 1520-1529.
- Duchaine TF, Slack FJ. RNA interference and micro RNA-oriented therapy in cancer: rationales, promises, and challenges. Curr Oncol. 2009; 16: 61-66.
- Misso G, Zappavigna S, Castellano M, De Rosa G, Di Martino MT, Tagliaferri P, et al. Emerging pathways as individualized therapeutic target of multiple myeloma. Expert Opin Biol Ther. 2013; 13 Suppl 1: S95-109.
- Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci U S A. 2008; 105: 5166-5171.
- Roccaro AM, Sacco A, Thompson B, Leleu X, Azab AK, Azab F, et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. 2009; 113: 6669-6680.
- Sun CY, She XM, Qin Y, Chu ZB, Chen L, Ai LS, et al. L miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis. 2013; 34: 426-435.
- Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013; 123: 1542-1555.
- Amodio N, Di Martino MT, Foresta U, Leone E, Lionetti M, Leotta M, Gulla AM, Pitari MR, Conforti F, Rossi M, Agosti V, Fulciniti M, Misso G, Morabito F, Ferrarini M, Neri A, Caraglia M, Munshi NC, Anderson KC, Tagliaferri P, Tassone P (2012) miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell death & disease 3:e436. doi:10.1038/cddis.2012.175
- Zhang YK, Wang H, Leng Y, Li ZL, Yang YF, Xiao FJ, et al. Over expression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem Biophys Res Commun. 2011; 414: 233-239
- Amodio N, Leotta M, Bellizzi D, Di Martino MT, D'Aquila P, Lionetti M, et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget. 2012; 3: 1246-1258.
- Amodio N, Bellizzi D, Leotta M, Raimondi L, Biamonte L, D'Aquila P, Di Martino MT, Calimeri T, Rossi M, Lionetti M, Leone E, Passarino G, Neri A, Giordano A, Tagliaferri P, Tassone P (2013) miR-29b induces SOCS-1 expression by promoter demethylation and negatively regulates migration of multiple myeloma and endothelial cells. Cell Cycle 12 (23):3650-3662. doi:10.4161/cc.26585
- Rossi M, Pitari MR, Amodio N, Di Martino MT, Conforti F, Leone E, Botta C, Paolino FM, Del Giudice T, Iuliano E, Caraglia M, Ferrarini M, Giordano A, Tagliaferri P, Tassone P (2012) miR-29b negatively regulates human osteoclastic cell differentiation and function: Implications for the treatment of multiple myeloma-related bone disease. Journal of cellular physiology. doi:10.1002/jcp.24306
- Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, Cantafio ME, Gulla A, Conforti F, Morelli E, Tomaino V, Rossi M, Negrini M, Ferrarini M, Caraglia M, Shammas MA, Munshi NC, Anderson KC, Neri A, Tagliaferri P, Tassone P (2012) Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clinical cancer research : an official journal of the American Association for Cancer Research 18 (22):6260-6270. doi:10.1158/1078-0432.CCR-12-1708
- Scognamiglio I, Di Martino MT, Campani V, Virgilio A, Galeone A, Gulla A, Gallo Cantafio ME, Misso G, Tagliaferri P, Tassone P, Caraglia M, De Rosa G (2014) Transferrin-conjugated SNALPs encapsulating 2'-O-methylated miR-34a for the treatment of multiple myeloma. BioMed research international 2014:217365. doi:10.1155/2014/217365
- Calimeri T, Battista E, Conforti F, Neri P, Di Martino MT, Rossi M, Foresta U, Piro E, Ferrara F, Amorosi A, Bahlis N, Anderson KC, Munshi N, Tagliaferri P, Causa F, Tassone P (2011) A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia 25 (4):707-711. doi:10.1038/leu.2010.300
- Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids. 2014; 3: e194.
- Di Martino MT, Campani V, Misso G, Gallo Cantafio ME, Gulla A, Foresta U, Guzzi PH, Castellano M, Grimaldi A, Gigantino V, Franco R, Lusa S, Cannataro M, Tagliaferri P, De Rosa G, Tassone P, Caraglia M (2014) In vivo activity of miR-34a mimics delivered by stable nucleic acid lipid particles (SNALPs) against multiple myeloma. PloS one 9 (2):e90005. doi:10.1371/journal.pone.0090005
- Raimondi L, Amodio N, Di Martino MT, Altomare E, Leotta M, Caracciolo D, Gulla A, Neri A, Taverna S, D'Aquila P, Alessandro R, Giordano A, Tagliaferri P, Tassone P (2014) Targeting of multiple myeloma-related angiogenesis by miR-199a-5p mimics: in vitro and in vivo anti-tumor activity. Oncotarget 5 (10):3039-3054
- Leone E, Morelli E, Di Martino MT, Amodio N, Foresta U, Gulla A, Rossi M, Neri A, Giordano A, Munshi NC, Anderson KC, Tagliaferri P, Tassone P (2013) Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clinical cancer research : an official journal of the American Association for Cancer Research. doi:10.1158/1078-0432.CCR-12-3325
- Di Martino MT, Gulla A, Gallo Cantafio ME, Altomare E, Amodio N, Leone E, Morelli E, Lio SG, Caracciolo D, Rossi M, Frandsen NM, Tagliaferri P, Tassone P (2014) In vitro and in vivo activity of a novel locked nucleic acid (LNA)-inhibitor-miR-221 against multiple myeloma cells. PloS one 9 (2):e89659. doi:10.1371/journal.pone.0089659
- Di Martino MT, Gullà A, Cantafio ME, Lionetti M, Leone E, Amodio N, et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget. 2013; 4: 242-255.
- Leotta , Biamonte L, Raimondi L, Ronchetti D, Di Martino MT, Botta C, et al. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J Cell Physiol. 2014; 229: 2106-2116.
- Rossi M, Di Martino MT, Morelli E, Leotta M, Rizzo A, Grimaldi A, et al. Molecular targets for the treatment of multiple myeloma. Curr Cancer Drug Targets. 2012; 12: 757-767.
- Neri P, Tagliaferri P, Di Martino MT, Calimeri T, Amodio N, Bulotta A, Ventura M, Eramo PO, Viscomi C, Arbitrio M, Rossi M, Caraglia M, Munshi NC, Anderson KC, Tassone P (2008) In vivo anti-myeloma activity and modulation of gene expression profile induced by valproic acid, a histone deacetylase inhibitor. British journal of haematology 143 (4):520-531. doi:10.1111/j.1365-2141.2008.07387.x
- Strausberg RL, Simpson AJ. Whole-genome cancer analysis as an approach to deeper understanding of tumour biology. Br J Cancer. 2010; 102: 243-248.
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, Cook L, Abbott R, Larson DE, Koboldt DC, Pohl C, Smith S, Hawkins A, Abbott S, Locke D, Hillier LW, Miner T, Fulton L, Magrini V, Wylie T, Glasscock J, Conyers J, Sander N, Shi X, Osborne JR, Minx P, Gordon D, Chinwalla A, Zhao Y, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson M, Baty J, Ivanovich J, Heath S, Shannon WD, Nagarajan R, Walter MJ, Link DC, Graubert TA, DiPersio JF, Wilson RK (2008) DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456 (7218):66-72. doi:10.1038/nature07485
- Kristensen VN, Lingjærde OC, Russnes HG, Vollan HK, Frigessi A, Børresen-Dale AL. Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 2014; 14: 299-313.
- Venkatesh TV, Harlow HB. Integromics: challenges in data integration. Genome Biol. 2002; 3: REPORTS4027.
- Camps C, Saini HK, Mole DR, Choudhry H, Reczko M, Guerra-Assuncao JA, Tian YM, Buffa FM, Harris AL, Hatzigeorgiou AG, Enright AJ, Ragoussis J (2014) Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Molecular cancer 13:28. doi:10.1186/1476-4598-13-28
- Kim S, Park T, Kon M (2014) Cancer survival classification using integrated data sets and intermediate information. Artificial intelligence in medicine 62 (1):23-31. doi:10.1016/j.artmed.2014.06.003
- Gade S, Porzelius C, Falth M, Brase JC, Wuttig D, Kuner R, Binder H, Sultmann H, Beissbarth T (2011) Graph based fusion of miRNA and mRNA expression data improves clinical outcome prediction in prostate cancer. BMC bioinformatics 12:488. doi:10.1186/1471-2105-12-488
- Yan Z, Shah PK, Amin SB, Samur MK, Huang N, Wang X, Misra V, Ji H, Gabuzda D, Li C (2012) Integrative analysis of gene and miRNA expression profiles with transcription factor-miRNA feed-forward loops identifies regulators in human cancers. Nucleic acids research 40 (17):e135. doi:10.1093/nar/gks395
- Bisognin A, Sales G, Coppe A, Bortoluzzi S, Romualdi C (2012) MAGIA(2): from miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update). Nucleic acids research 40 (Web Server issue):W13-21. doi:10.1093/nar/gks460
- Huang GT, Athanassiou C, Benos PV. mirConnX: condition-specific mRNA-microRNA network integrator. Nucleic Acids Res. 2011; 39: W416-423.
- Huang JC, Babak T, Corson TW, Chua G, Khan S, Gallie BL, et al. Using expression profiling data to identify human microRNA targets. Nat Methods. 2007; 4: 1045-1049.
- Ruike Y, Ichimura A, Tsuchiya S, Shimizu K, Kunimoto R, Okuno Y, et al. Global correlation analysis for micro-RNA and mRNA expression profiles in human cell lines. J Hum Genet. 2008; 53: 515-523.
- Gutierrez NC, Sarasquete ME, Misiewicz-Krzeminska I, Delgado M, De Las Rivas J, Ticona FV, Ferminan E, Martin-Jimenez P, Chillon C, Risueno A, Hernandez JM, Garcia-Sanz R, Gonzalez M, San Miguel JF (2010) Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 24 (3):629-637. doi:10.1038/leu.2009.274
- Wang L, Oberg AL, Asmann YW, Sicotte H, McDonnell SK, Riska SM, Liu W, Steer CJ, Subramanian S, Cunningham JM, Cerhan JR, Thibodeau SN (2009) Genome-wide transcriptional profiling reveals microRNA-correlated genes and biological processes in human lymphoblastoid cell lines. PloS one 4 (6):e5878. doi:10.1371/journal.pone.0005878
- Zhang Y, Liu W, Xu Y, Li C, Wang Y, Yang H, Zhang C, Su F, Li Y, Li X Identification of Subtype Specific miRNA-mRNA Functional Regulatory Modules in Matched miRNA-mRNA Expression Data: Multiple Myeloma as a Case. BioMed research international