
Case Report
Ann Hematol Oncol. 2015;2(6): 1050.
Acute Myeloid Leukemia with t(11;17)(q23;q21)
Piñán MA*, Balerdi A, Iglesias A, Dueñas M, Olazabal I, Puente M, Amutio E, Del Orbe R, Ancin I and García-Ruiz JC
Department of Haematology, Cruces University Hospital, Vizcaya, Spain
*Corresponding author: Maria Angeles Piñan, Department of Haematology, Cruces University Hospital, Spain
Received: July 14, 2015; Accepted: August 10, 2015; Published: August 12, 2015
Abstract
Acute promyelocytic leukemia (APL) is characterized by the presence of t(15;17), which generates the PML-RARα transcript and by a good response to treatment with retinoids; However, in some variant types as APL with t(11;17) this benefit is not well established. We present the case of a patient who received chemotherapy associated with acid trans-retinoic (ATRA) who achieved complete molecular response and a long term follow-up survival.
Keywords: APL; PLZF-RARα
Introduction
Acute myeloid leukemia with t(11;17)(q23;q21) is are variant of APL, with morphologic characteristics that differentiates from the classic form and in which there is little experience of treatment.
Case Presentation
A woman of 49 years old, diagnosed with rheumatoid arthritis in January 2011 and undergone to treatment with corticosteroids, methotrexate and hydroxychloroquine was controlled since March 2012 by leukopenia.
On May 31st of 2012 she consulted for presenting anemia (Hb: 97g/L), leukopenia (1.16 x 109L) and neutropenia (0.2 x 109L), attributed to the treatment with methotrexate, which was suspended and instead, a dose of granulocyte colony stimulating factors (G-CSF) was given to the patient; 24 hours later she developed abdominal pain, nausea, vomits, hematuria and oliguria. Three days later she failed on a relevant renal insufficiency (creatinine: 7.6 mg/dl), deeper anemia and thrombocytopenia. She was treated with hemodialysis, transfusions of red blood cells and platelets and steroids. Nine days later she transferred to our hospital with suspicion Thrombotic Thrmbocitopenic Purpura, so that dialysis and plasma transfusions was rebooted.
Physical examination, abdominal ultrasonography and renal Doppler, showed no abnormalities.
Biochemical profile: Urea: 145 mg/dL (10-50), Creatinine: 4.61 mg/dL (0.1-1.1), LDH: 422 u/L (26-245) and rest of parameters were within normal limits.
Blood test revealed: Hb: 82 g/L, MCV: 94.6 fl, WBC: 7.9 x 109/L (52% of cells in medium size, round nucleus and pseudo-pelger with wide cytoplasm and fine granulation), Platelets: 92 x 109/L (Figure1). Erythrocyte morphology: 2 schistocytes/1000 erythrocytes (Figures 1,2).
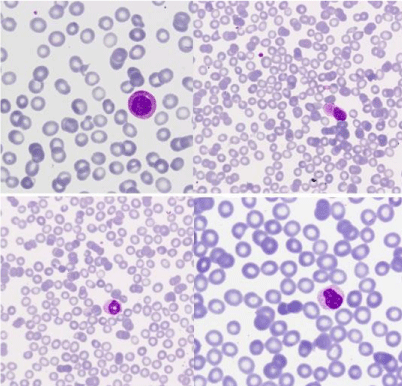
Figure 1: Images of peripheral blood: granulocytic dysplastic features
(nuclear ring, chromatin with crumpling phenomena) and atypical cells
of eccentric nucleus and large cytoplasm with fine granulation and polar
distribution. (MGGx1000).
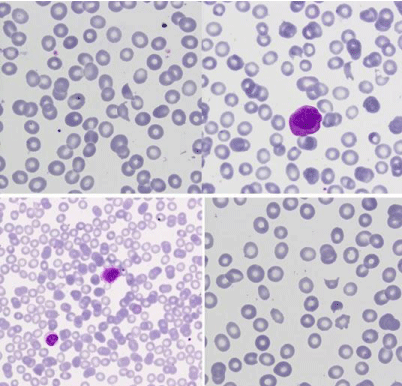
Figure 2: Peripheral blood: granular blasts and isolated schistocytes
(MGGx1000).
Coagulation test: Kaolin cephalin clotting time: 32” (24-35), Prothrombin activity: 66% (70-120), Fibrinogen: 236 mg/dL (200- 400), D-dimers: 11880 ng/mL (0-500), Fibrin monomers: absent.
Bone marrow aspiration (BMA) showed a hipercellular image, with 71% of cells similar as the peripheral blood; no faggot cells or Auer rods were seen and reaction with peroxidase was strong. (Figure 3-9).
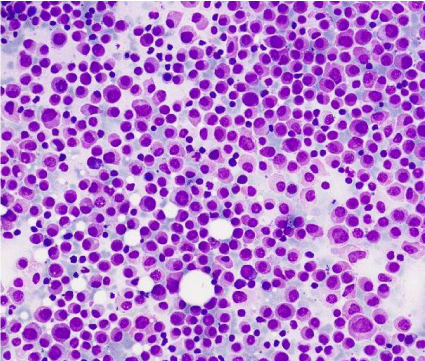
Figure 3: Bone marrow aspirate which shows an hypercellular and
monomorphic image, constituted by a blast population of medium size, with a
round eccentric nucleus and a large cytoplasm with fine granulation, without
Auer rods (MGGx200).
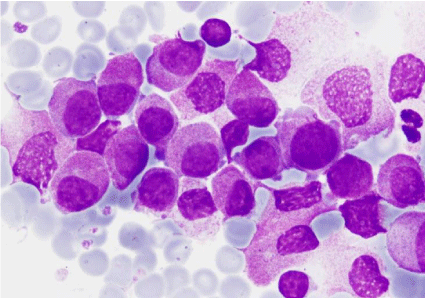
Figure 4: Bone marrow aspirate: blasts (MGGx1000).
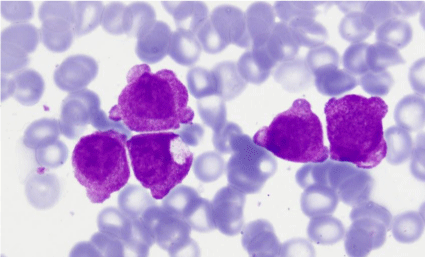
Figure 5: Bone marrow aspirate: granular blasts with presence of blebs in
their outline (MGGx1000).
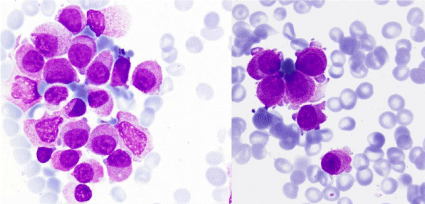
Figure 6: Bone marrow aspirate: blasts with a mature appearance nucleus
and great fragility (MGGx1000).
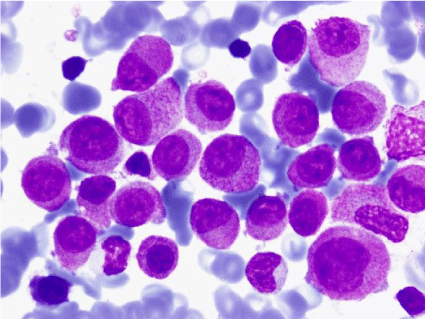
Figure 7: Bone marrow aspirate: blasts and an erythroblast with lackluster
chromatin (MGGx1000).
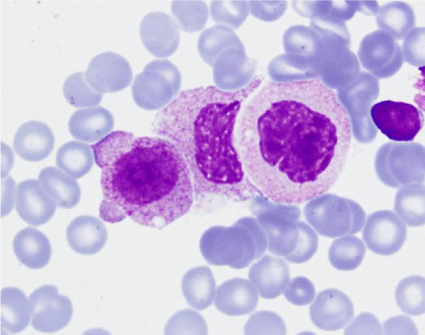
Figure 8: Bone marrow aspirate: blast with blebs and probably degranulated
cells (MGGx1000).
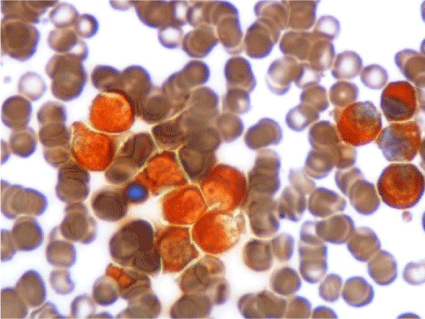
Figure 9: Bone marrow aspirate: strong myeloperoxidase activity (Px1000).
Immunophenotyping: these cells were: CD45 +, MPO +, CD13 +, CD33 +, CD117 +, DR-, CD34-, CD56 +, CD11c +, CD32 +, CD64 +, CD38 +, CD11b-, CD16-, CD14-, CD4- (Figure 10).
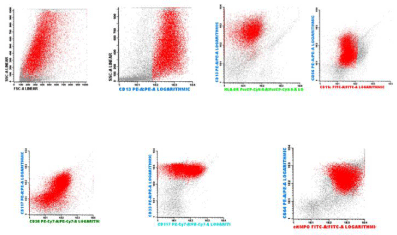
Figure 10: Inmunophenotype on bone marrow: the blast population was
characterized by markers CD45 +, + CD13, CD33 +, CD117 + DR - CD56 +
CD11c +, CD32 +, CD64 +, CD38 +, MPO +, CD34-, CD11b-, CD16-, CD14-,
CD4 -.
The cytogenetics and molecular studies on BM were:
- Rearrangement PML-RARα (q21; q22) t(15;17) (RT-PCR): negative.
- FISH: nucish 15q22 (PMLx2), 17q21 (RARax3) [23/100] (Figure 11).
- Karyotype: 46, XX, del(5)(q13q31), t(11;17)(q23;q21) [20/30] / 46, XX [10/30] (Figure 12).
- Rearrangement PLZF-RARα positive (RT-PCR) (Figure 13).
- Rearrangement RARα-PLZF positive (RT-PCR).
- Jovanovic JV, Rennie K, Culligan D, Peniket A, Lennard A, Harrison J et al. Development of real-time quantitative polymerase chain reaction assays to track treatment response in retinoid resistant acute promyelocyticleukemia. Frontiers in oncology. 2011; 35: 1-8.
- Suliman BA, Xu D, Williams BR. The promyelocytic leukemia zinc finger protein: two decades of molecular oncology. Front Oncol. 2012; 2: 74.
- Guidez F, Parks S, Wong H, Jovanovic JV, Mays A, Gilkes AF, et al. RARalpha-PLZF overcomes PLZF-mediated repression of CRABPI, contributing to retinoid resistance in t(11;17) acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2007; 104: 18694-18699.
- Grimwade D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M et al. On behalf of Group e Francais de Cytogénétique Hematologique, Groupe Francaisd’ Hematologie Cellulaire, UK Cancer Cytogenetics Group, and BIOMED 1 European Community-Concerted. Action “Molecular Cytogenetic Diagnosis in Haematological Malignancies” Characterization of acute promyelocyticleukemia cases lacking the classic t(15;17): results of the European Working Party. Blood. 2000; 96: 1297-1308.
- Rohr SS, Flores Pelloso LA, Borgo A, De Nadai LC, Yamamoto M, Rego EM et al. Acute promyelocyticleukemia associated with the PLZF-RARA fusion gene: two additional cases with clinical and laboratorial peculiar presentations. Med Oncol. 2011; 266-271.
- Cassinat B, Guillemot I, Moluçon-Chabrot C, Zassadowski F, Fenaux P, Tournilhac O, et al. Favourable outcome in an APL patient with PLZF/RARalpha fusion gene: quantitative real-time RT-PCR confirms molecular response. Haematologica. 2006; 91: ECR58.
- Licht JD, Chomienne C, Goy A, Chen A, Scott AA, Head DR, et al. Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood. 1995; 85: 1083-1094.
- Jansen JH, de Ridder MC, Geertsma WM, Erpelinck CA, van Lom K, Smit EM, et al. Complete remission of t(11;17) positive acute promyelocytic leukemia induced by all-trans retinoic acid and granulocyte colony-stimulating factor. Blood. 1999; 94: 39-45.
- Ramadan SM, Fouad TM, Summa V, Hasan SKh, Lo-Coco F. Acute myeloid leukemia developing in patients with autoimmune diseases. Haematologica. 2012; 97: 805-817.
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 2008; Vol 2. Geneva (Switzerland) WHO Press.
- Dong HY, Kung JX, Bhardwaj V, McGill J. Flow cytometry rapidly identifies all acute promyelocytic leukemias with high specificity independent of underlying cytogenetic abnormalities. Am J Clin Pathol. 2011; 135: 76-84.
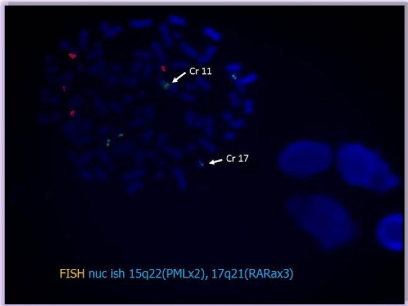
Figure 11: Technique of FISH on the bone marrow aspirate showing a signal
on chromosome 11.
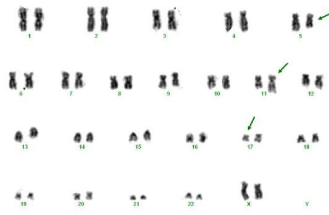
Figure 12: Cytogenetics of BM which confirms the t(11;17))(q23;q21), in
addition to one of the (5q) (q13q31) in 20/30 metaphases.
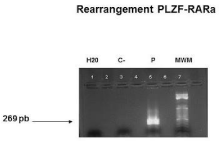
Figure 13: Rearrangement PLZF-RARa: RT-PCR that demonstrates the
presence of a band of 269bp; C-: negative control, P:patient, MPM: molecular
weights marker.
Diagnostic: Acute Promyelocytic Leukemia with variant translocation RARα (WHO classification).
Follow up: on 6/28/2013 the patient began induction chemotherapy (Idarubicin, 12 mgr/m2/d on days 1, 3 and 5; ARA-C, 500 mg/m2twice daily on days, 1, 3, 5 and 7 and Etoposide, 100 mg/ m2/d on days 1 through 3) + ATRA, 45 mg/m2day divided into 2 doses till achieving complete remission).
BMA on day + 37 post-induction showed a 3% blast cells, residual disease (MRD) by flow cytometry of 0.4% and a karyotype: 46, XX, del(5)(q13q31), t(11;17)(q23;q21) [4/50] / 46, XX [46/50]; and PLZFRARα rearrangement was positive.
After that treatment of consolidation was started (2 cycles of ARA-C, 500 mg/m2 / 12 h, on day 1 to 6; Mitoxantrone 12 mg/m2/d on days4, 5 and 6 and ATRA, 45 mg/m2/d divided into two doses, on day 1 to 15, after which normalized the karyotype, the MRD: 0.072% and the PLZF-RARα and RARα-PLZF rearrangements were negative.
In February 2013 allogeneic hematopoietic stem cell transplantation (HSCT) obtained from peripheral blood of her sister with myeloablative conditioning was done, without significant adverse events.
Discussion
APL is included among leukemias with recurrent cytogenetic anomalies and characterized by translocation t(15;17)(q22;q12) PMLRARα. However, in 10%of cases, this alteration is not detected by cytogenetic techniques, either by technical problems, submicroscopic chromosomal insertions or more complex rearrangements. In spite of this, an accurate diagnosis can be done since the PML-RARa transcript would be positive.
In distinction to, approximately 2% of cases of APL carry a fusion gene alternative to the PML (PLZF with t(11;17)(q23q21), NPM1 with t(5;17), NuMA with t(11;17)(q13q21), FIPIL1 with the t(4;17), BCORSTAT5b , the t(X;17), PRKAR1A with rearrangements of 17q).
The most frequent of all of them is the PLZF (promielocytic zinc finger) [1]; this gene also known as Zbtb16 was described at first time in 1993 in a Chinese patient who suffered an atypical APL; It’s located on chromosome 11q23 among a cluster of family zinc finger genes, coding of proteins with high content of cysteine and histidine that require one or more unions of zinc to stabilize its structure [2]; It has a high capacity repressor of transcription and his expression is very high in stem cells and progenitor, but decreases as cells get madure.
The clinical relevance of these variants of APL is without a doubt the different response to treatment with ATRA and Arsenic Trioxide, due to all of them are sensitive with the exception of carriers of the rearrangements with PLZF and STAB5b genes.
In cases of APL with t(11;17)(q23q21) there is evidence of this resistance both in vivo as in vitro, particularly in those patients who have the reverse rearrangement RARα-PLZF, whose product would play an important role in mediating resistance to ATRA [1,3].
Since 1993 there have been reported 16 cases of patients with this variant translocation [4-7], with a very low incidence in women (14 men versus two women) and frequent coagulopathy (11 patients). t(11;17) was detected in all of them by conventional cytogenetics or by FISH, and molecular assays was positive to the PLZF-RARα and RARα-PLZF rearrangements. According to treatment, 14 received ATRA, 13 with concomitant chemotherapy following different schemes; 4 were undergone to Allo-HSCT and one to Auto-HSCT, getting responses of variable duration in all patients.
In our case disease onset was peculiar, with “sub-acute” presentation, absence of hemorrhagic diathesis and acute renal failure.
Relationship between treatment with stimulating factors and kidney damage lead us to think that the pro-inflammatory or thrombotic effects of the G-CSF could play some role. However, it was not demonstrated the existence of a renal thrombosis, or data that support a clear endothelial damage. In the other hand one reported case was treated with the association of ATRA plus G-CSF with good results [8] and our patient resolved her kidney disease with chemotherapy.
15 days before to diagnosis of APL the patient received multiple plasma transfusions and dialysis that could have masked hemostasis alterations in this type of leukemia; even so, we detected signs of fibrinolysis days prior to the treatment.
A history of rheumatoid arthritis treated with Methotrexate and presence of a del5q in the karyotype, allows us to speculate with the possible existence of a myelodysplasia prior to the development of leukemia or relate both diseases. In this regard, we do not have data that showing the first possibility.
Otherwise, majority of studies that have investigated the association of autoimmune diseases and their treatments with AML, have described an increase in the risk of developing it, although does not reach statistical significance, so considering these leukemia as related to therapy (t-AML) it does not seem appropriate [9].
In the PETHEMA (Spanish Protocol for the study of malignant hemopathies) registry, there are 1771 patients diagnosed with APL recorded with assessable cytogenetic or molecular studies from 1996 to 2015, of which only two patients presented the variant translocation t(11;17)(q23; q21)(the present case and other), representing the 0.1% of the total. Both of them were treated with chemotherapy plus ATRA achieving a molecular complete response with a follow-up in our case of 38 months.
Morphological diagnosis of the variant APL´ is always challenging; in the literature many cases were described as an hybrid between AML-M2 and M3 (FAB classification) [7] and the blasts, were defined as cells blocked at an intermediate stage promyelocytemyelocyte, with a nucleus of mature appearance and condensed chromatin, as well as presence of dysplastic traits associated (hypo granulation, nuclear pseudo pelger). Description of the blasts which makes the WHO classification of 2008 does not differ much from these initial descriptions [10].
However, immunophenotypic profile of APL is independent of underlying cytogenetic variations, a quick and essential tool for the diagnosis [11] which of course must be completed with the cytogenetic and molecular studies.
References