
Case Report
Ann Hematol Oncol. 2015; 2(9): 1062.
Dual Diagnosis of Multiple Myeloma and T-cell Large Granular Lymphocytic Leukemia: A Case Report and Literature Review
Carreau NA¹, Lee JY², Petersen B¹, Silverman L¹ and Chari A¹*
¹Mount Sinai Medical Center, USA
²Columbia University Medical Center, USA
*Corresponding author: Ajai Chari, Affiliated with Mount Sinai Medical Center, 1 Gustave Levy Place, Box 1185, New York, NY 10029, USA
Received: November 03, 2015; Accepted: December 10, 2015; Published: December 12, 2015
Abstract
A 52 year-old Caucasian male with stable Crohn’s disease off medications, with anemia and known vitamin B12 and iron deficiency, was noted to be pancytopenic with a white blood cell count of 1,600/uL and a platelet count of 106,000/uL. He also noted night sweats, fatigue, and a distended abdomen. While admitted with febrile neutropenia due to methicillin-susceptible Staphylococcus aureus (MSSA) bacteremia in the setting of a rectal abscess, further workup revealed a new diagnosis of lambda light chain multiple myeloma (MM). After 4 cycles of chemotherapy, he achieved a partial response but remained cytopenic and developed increased splenomegaly. Extensive workup resulted in the diagnosis of T-cell large granular lymphocytic leukemia (T-LGLL) by T-cell receptor gene rearrangement studies. Initiation of oral cyclophosphamide with corticosteroids resulted in improvement in blood counts, and he was consolidated with an allogeneic stem cell transplant. His MM appeared to be in a stringent complete remission at 30 months, whereas the T cell rearrangement for T-LGLL was initially negative but then became positive again 7 months later.
T –LGLL is a clonal disease of large granular lymphocytes, typically cytotoxic T cells, which accounts for 2-3% of all cases of chronic lymphoproliferative disorders. Common symptoms include fever, recurrent bacterial infections, weight loss, and fatigue. Labs usually note cytopenias, and both splenic and hepatic involvement are common. T-LGLL is often associated with autoimmune disorders, and may co-exist with other hematologic malignancies, seldomly with a primary diagnosis of multiple myeloma (MM). Given the rarity of the disease and its typically indolent course, there are no standard treatment guidelines, and immunosuppressive chemotherapy is reserved for cases of symptomatic cytopenias and splenomegaly. In an era with increasing concerns about secondary malignancies in MM, the differential for patients with persistent cytopenias and/or splenomegaly despite treatment should include concomitant T-LGLL.
Keywords: T-LGLL; Multiple myeloma; Splenomegaly; Cytopenia
Case Presentation
A 52 year-old Caucasian male with Crohn’s disease stable off medications for several years, with anemia and known vitamin B12 and iron deficiency, was noted to be pancytopenic with a white blood cell (WBC) count of 1,600/uL and a platelet count of 106,000/uL for two months. The patient endorsed night sweats over the preceding two years, a distended abdomen for six months, and recent fatigue. He then developed 15 days of fever and was found to be neutropenic with methicillin-susceptible Staphylococcus aureus (MSSA) bacteremia in the setting of a rectal abscess.
Initial labs revealed a WBC count of 1,200/uL, with only 6% segmented cells, 38% lymphocytes, 26% eosinophils, 12% monocytes and 16% basophils, with an absolute neutrophil count of 72/uL without bands. Hemoglobin was 8.9 g/dL (MCV 80 fl), platelet count241,000/ uL, creatinine 0.79 mg/dL, calcium 8.5/mL, total protein 5.4 mg/dL, albumin 2.6 mg/dL, and beta-2-microglobulin 4.51 mg/L.ESR was elevated to 87 mm/hr and CRP 28.1 mg/L. Chemistry revealed mild hypokalemia, and hepatic function tests were normal.
The Immunoglobulin G (IgG) and IgA were within normal limits at 1066 mg/dL and 342 mg/dL respectively, but IgM was low at 6 mg/ dL (normal 40-320). Serum protein electrophoresis (SPEP) revealed two monoclonal bands in the gamma region too low to quantitate. Serum immunofixation electrophoresis (SIFE) revealed a free monoclonal lambda light chain and faint IgG lambda band. 24h urine protein electrophoresis (UPEP) revealed 12.4 g proteinuria with 10.1 g Bence Jones protein (BJP) identified on urine immunofixation electrophoresis (UIFE) as lambda light chains. Serum free kappa chains were 25.05 mg/L and serum free lambda chains were 2034 mg/L (normal 5.71-26.30 mg/L), with a kappa: lambda ratio of 0.012 (normal 0.26-1.65).
Initial bone marrow (BM) biopsy revealed 10% lambda-restricted plasma cells. Repeat biopsy showed a mildly hypercellular marrow with patchy infiltrates of lambda light chain restricted CD 138 positive plasma cells comprising 20% of the bone marrow cellularity, consistent with lambda light chain MM. Cytogenetics and FISH were normal.
Positron emission tomography-computed tomography (PETCT) revealed diffuse uptake in the BM of the axial and appendicular skeleton without focal or osteolytic lesions, in addition to bilateral hypermetabolic inguinal, axillary, and mesenteric lymph nodes up to 1.2 cm in size. Hepatomegaly at 22 x 16 cm and splenomegaly at 20 cm in long axis without abnormal FDG uptake were also seen. The patient was diagnosed with lambda light chain multiple myeloma, Durie-Salmon Stage 3A due to nadir Hg 7.4 and International Staging System (ISS) Stage 2.
Bortezomib was given subcutaneously on days 1,4,8, and 11 along with dexamethasone 40 mg orally on the same days. Thalidomide 100mg then 200mg was added to cycles 2 through 4 with a partial response (PR). Due to sensory neuropathy, thalidomide was replaced with cyclophosphamide 250mg/m2 IV (dose reduced for cytopenias) on days 1 and 8. After a total of 5 cycles of chemotherapy, 24hour UPEP revealed a 61.5% reduction in BJP consistent with stable disease (SD).
BM biopsy was repeated to re-evaluate myelosuppression given the need for granulocyte colony-stimulating factor (G-CSF) support throughout induction chemotherapy with a relatively non-myelosuppresive regimen. This revealed a hypercellular (70%) marrow with trilineage hematopoiesis showing full maturation, with 10-20% plasma cells and no evidence of myelodysplasia. Cytogenetics and flow cytometry were unrevealing.
The patient suffered a subacute ischemic frontoparietal stroke with a platelet count of 60,000/uL, and subsequent paroxysmal nocturnal hemoglobinuria testing was negative. Repeat PET-CT revealed increased splenomegaly to 22cm x 13cm (Figure 1). Testing for Gaucher’s disease, Felty’s syndrome (with rheumatoid factor), autoimmune disease (with antinuclear antibodies), and amyloid (with congo red stain of BM, fat pad, liver and rectum) was negative. G-CSF and plerixafor based stem cell harvest was attempted twice with a combined yield of only 2.37* 10^6 CD34+/kg.
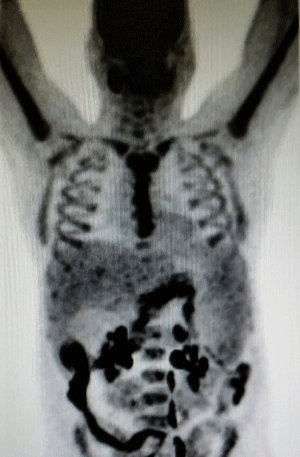
Figure 1: Hepatosplenomegaly as evidenced on PET-CT.
Doppler ultrasound of the liver ruled out portal hypertension to explain the splenomegaly. Transjugular liver biopsy showed extensive extramedullary hematopoiesis in the sinusoids but no signs of lymphoma. Repeat BM biopsy showed increased hypercellularity of 90%, with patchy infiltrates of lambda-restricted plasma cells (20- 30% overall cellularity), with no evidence of myelodysplasia. Due to the progression of MM, dexamethasone 40 mg twice weekly was started with SD.
Polymerase chain reaction (PCR) was done to test for clonal T and B cell receptor rearrangement, which revealed a clonal T cell receptor beta gene rearrangement in the V and J regions, consistent with a monoclonal and/or oligoclonal T cell population. There was no gamma arrangement. Re-evaluation of the liver biopsy revealed an intrasinusoidal infiltrate of small atypical T-lymphocytes; CD3+, CD7+, CD5-, CD8+, CD4-, CD56-, CD57-, by immunoperoxidase stains. The patient was diagnosed with T-LGLL and started on oral cyclophosphamide 50 mg bid in addition to prednisone and clarithromycin, to potentiate the steroid anti-myeloma effect. Although the MM remained stable, blood counts notably improved. WBC rose from 1,300/uL to 9,200/uL and platelet count increased from 60,000/uLto 121,000/uL after the initiation of the treatment. He was then referred for transplant at an outside hospital.
The patient underwent allogeneic peripheral blood stem cell transplant (allo SCT) from a 10/10 HLA matched, ABO major incompatible, CMV positive male unrelated donor using Melphalan, Fludarabine, and rabbit Antithymocyte globulin conditioning. Transplant course was complicated by a Crohn’s flare, Escherichia coli sepsis, and febrile neutropenia. Neutrophil and platelet engraftment occurred on day 16 post-transplant. He received a short course of methotrexate and was continued on tacrolimus for immunosuppression.
Post-transplant course was complicated by respiratory syncytial virus infection and acute graft versus host disease (GVHD) of the skin. He subsequently developed a parvovirus infection, which was treated with intravenous immunoglobulin (IVIG) and Rituximab. On day83 post-allo SCT, the MM appeared to be in a stringent complete remission with no evidence of disease in blood, urine, or BM; and T cell rearrangement for T-LGLL was negative. However 7 months after transplant the T cell clone was again detected by PCR, while blood counts remained stable. 30 months post-transplant, the patient’s MM remains in remission.
Discussion
T-cell large granular lymphocytic leukemia (T–LGLL) is a clonal disease of large granular lymphocytes, typically cytotoxic T cells, accounting for 2-3% of cases of chronic lymphoproliferative disorders [1]. The median age at diagnosis is 60, with both men and women affected equally [2,3]. It is typically an indolent disease, with a median survival of ten years [3].
Symptoms of T-LGLL include fever, recurrent bacterial infections, weight loss, and fatigue [3,4]. Clinical signs include cytopenias. Thrombocytopenia is usually of moderate severity, and can be associated with antiplatelet antibodies, making it difficult to differentiate from immune thrombocytopenia (ITP) [3,5]. Splenic involvement is common (20-50%), in addition to hepatic involvement with portal and sinusoidal infiltration [6]. Patients commonly have autoimmune disorders, most often rheumatoid arthritis. T-LGLL can also be concomitant with other hematologic disorders such as ITP, aplastic anemia, pernicious anemia, and other malignancies [7,8]. There is a single case report of a patient a with T-LGLL who then developed Crohn’s disease, however it is well-established that inflammatory bowel disease and T-LGLL are associated, likely due to an autoimmune diathesis [4,9-11].
Our patient initially presented with MSSA bacteremia in the setting of three months of pancytopenia with initial BM biopsy showing a low burden of 20% plasma cells. There was an initial partial response with bortezomib and thalidomide treatment, but further treatments were complicated by recurrent cytopenias despite a relatively non-myelosuppressive regimen. The patient also failed two stem cell harvests with low yield, and his splenomegaly actually worsened amidst treatment for MM. Despite extensive testing it was only with T-cell receptor gene rearrangement studies that T-LGLL was diagnosed.
T-LGLL is known to coexist with other lymphoid or myeloid clonal hematologic malignancies, but this case is uncommon in that typically the T-LGLL is diagnosed first or the primary malignancy has already been well established. T-LGLL has been seen with chronic lymphocytic leukemia, follicular lymphoma, hairy cell leukemia, mantle cell lymphoma, Hodgkin lymphoma, and rarely MM [7,8,12]. Clonal B cell dyscrasias may be seen in up to 25% of patients with LGL leukemia [13]. Although its etiology remains unknown, chronic immune stimulation is hypothesized to play a role.
The diagnosis of T-LGLL should be suspected in patients with cytopenias and LGLs seen on peripheral blood smear. The WHO diagnosis of T-LGLL is delineated by greater than six months of increased LGL (>2 x 109/L) based on cytomorphology and flow cytometry, which reveals expansion of the cytotoxic CD8+ T cells [3,5,7]. T-cell receptor gene rearrangement PCR analysis is the most commonly used method to establish clonality in the LGL disorders, although the sensitivity is only 70-80% [3,4,14]. Peripheral blood smear will reveal characteristic large sized cells with a round nucleus and abundant cytoplasm [7,15], and BM biopsy typically demonstrates mild hypercellularity [8,13].
Although typically an indolent disease with favorable prognosis, indications for treatment include symptomatic cytopenias and progressive splenomegaly [7,16], as seen in our patient. Given its rarity, there are no standard guidelines, and therapy is based upon case reports and small studies [7,17]. Methotrexate, cyclosporine, cyclophosphamide, and prednisone have been used with varied success rates.
References
- McKenna RW, Parkin J, Kersey JH, Gajl-Peczalska KJ, Peterson L, Brunning RD. Chronic lymphoproliferative disorder with unusual clinical, morphologic, ultrastructural and membrane surface marker characteristics. Am J Med. 1977; 62: 588-596.
- Lamy T, Loughran TP Jr. Clinical features of large granular lymphocyte leukemia. Semin Hematol. 2003; 40: 185-195.
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist. 2004; 9: 247-258.
- O'Malley DP. T-cell large granular leukemia and related proliferations. Am J Clin Pathol. 2007; 127: 850-859.
- Loughran TP Jr, Clark EA, Price TH, Hammond WP. Adult-onset cyclic neutropenia is associated with increased large granular lymphocytes. Blood. 1986; 68: 1082-1087.
- Lamy T, Loughran TP Jr. Current concepts: large granular lymphocyte leukemia. Blood Rev. 1999; 13: 230-240.
- Prochorec-Sobieszek M. Advances in diagnosis and treatment of large granular lymphocyte syndrome. Curr Opin Hematol. 2011; 18: 55-62.
- Pontikoglou C, Kalpadakis C, Papadaki HA. Pathophysiologic mechanisms and management of neutropenia associated with large granular lymphocytic leukemia. Expert Rev Hematol. 2011; 4: 317-328.
- Kondoh K, Morimoto M, Keino D, Oyama R, Nagae C, Ashikaga T, et al. T-cell large granular lymphocyte leukemia in a child with anemia and Crohn's disease. Pediatr Int. 2013; 55: 111-114.
- Hoffman R, Benz E, Silberstein L, Heslop H, Weitz J, Anastasi J. Chapter 30: Acquired Disorders of Red Cell, White Cell, and Platelet Production. In: Hematology: Basic Principles and Practice, 6th Edition. Philadelphia, PA. 2013; 395: 19103-2899.
- Kondo H, Watanabe J, Iwasaki H. T-large granular lymphocyte leukemia accompanied by an increase of natural killer cells (CD3-) and associated with ulcerative colitis and autoimmune hepatitis. Leuk Lymphoma. 2001; 41: 207-212.
- Rossi D, Franceschetti S, Capello D, De Paoli L, Lunghi M, Conconi A, et al. Transient monoclonal expansion of CD8+/CD57+ T-cell large granular lymphocytes after primary cytomegalovirus infection. Am J Hematol. 2007; 82: 1103-1105.
- Evans HL, Burks E, Viswanatha D, Larson RS. Utility of immunohistochemistry in bone marrow evaluation of T-lineage large granular lymphocyte leukemia. Hum Pathol. 2000; 31: 1266-1273.
- Ryan DK, Alexander HD, Morris TC. Routine diagnosis of large granular lymphocytic leukaemia by Southern blot and polymerase chain reaction analysis of clonal T cell receptor gene rearrangement. Mol Pathol. 1997; 50: 77-81.
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program. 2012; 2012: 652-659.
- Hodges E, Krishna MT, Pickard C, Smith JL. Diagnostic role of tests for T cell receptor (TCR) genes. J Clin Pathol. 2003; 56: 1-11.
- Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood. 2011; 117: 2764-2774.