
Case Report
Ann Hematol Oncol. 2015; 2(10): 1068.
The Case of a Late Bleeder Plasminogen Activator Inhibitor 1 Deficiency
Del Rosario M¹* and Tsai H1,2
¹Eisenhower Medical Center, USA
²Luci Curci Cancer Center, USA
*Corresponding author: Michael Del Rosario, Eisenhower Medical Center, 39000 Bob Hope Dr, Rancho Mirage, CA 92270, USA
Received: September 23, 2015; Accepted: November 30, 2015; Published: December 02, 2015
Abstract
A 79-year-old post-menopausal Caucasian woman was investigated for lifelong history of bleeding including heavy menses, epistaxis, and minor postoperative bleeding such as post tonsillectomy and wisdom teeth extraction. Her history included significant hemorrhagic effusion post MRI scans of the right knee with no apparent cause for bleeding. Abnormalities and deficiencies of the coagulation system, platelet quantity/quality, alpha-2 plasmin inhibitor, and von Willebrand factor were negative. Patient was finally found to have heterozygous PAI-1 locus 4G, 5G deletion and insertion allele, demonstrating intermediate PAI-1 activity. This is striking, as normally severe PAI-1 deficiency is associated with recurrent bleeding episodes. These findings highlight the fact that even intermediate deficiency of the plasma PAI-1 can result in morbid clinical outcomes, therefore requiring the astute clinician to at least think about PAI-1 deficiency in a patient with recurrent bleeding episodes.
Keywords: Plasminogen Activator Inhibitor 1 Deficiency; Delayed Postoperative Bleeding; Bleeding Disorders; Coagulation Disorders; Alpha-2 Plasmin Inhibitor; Von Willebrand Factor Type 2N (Normandy); Hypofibrinogenemia; Dysfibrinogenemia
Background
Plasminogen Activator Inhibitor type 1 (PAI-1) is a little known but vital component of the body’s coagulation system. PAI- 1 is responsible for down regulating degradation of thrombi to help regulate fibrinolysis [1]. Normally, plasminogen activators circulate in plasma as a complex with PAI-1 as a reversible complex. When the fibrin clot is formed, plasminogen and t-PA bind to the surface of the clot. After protein interaction, plasmin is formed, which leads to fibrinolysis of the cross-linked fibrin resulting in fibrindegradation products. PAI-1 also binds to fibrin, and when bound, it can irreversibly bind to inhibitor t-PA (Figure 1) [2]. As one can expect, PAI-1 deficiency may end up as increased bleeding with up regulation of fibrinolysis. However, case reports of PAI-1 deficiency are rare. The first case report has been initially identified in the 1980s [3]. The affected patients express mild to moderate bleeding symptoms ranging from epistaxis to delayed bleeding after surgical procedures. Complete deficiency, however, is linked to life threatening hemorrhage and prolonged wound healing [4]. Aminocaproic acid or tranexamic acid are excellent fibrinolysis inhibiting drugs used as prophylaxis or treatment of bleeding episodes in patients with PAI-1 deficiency [5].
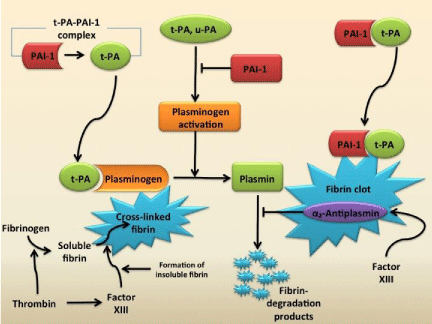
Figure 1: Activation and Inhibition of the Fibrinolytic Pathway.
Tissue plasminogen activator (t-PA) circulates in plasma as a complex
with plasminogen-activator inhibitor type 1 (PAI-1). Reactions occur on the
surface of the fibrin clot. t-PA or urinary-type plasminogen activator (u-PA)
activates plasminogen. A ternary complex of plasminogen, t-PA, and fibrin
promotes the formation of plasmin. Plasmin causes lysis of the cross-linked
fibrin into fibrin degradation products. PAI-1 also binds to fibrin and is able
to retain inhibition against t-PA. α2-antiplasmin inhibits plasmin from creating
fibrin degradation products. Factor XIII activates α2-antiplasmin as well as
cause soluble fibrin to become cross-linked fibrin. [2] Figure adapted from [2]
and created on Microsoft PowerPoint.
Case Presentation
“Ms. Y” is a 79-year-old, postmenopausal, Caucasian woman who has a history of degenerative arthritis and right knee pain for 15 years. Her pain became disabling in late summer of 2014. Ms. Y received two intra-Articular injections of 2% lidocaine and 40mg Kenalog in September 2014. There was 60% and 85% improvement in the pain after the first and second injections, respectively. On September 29, 2014, she presented to her primary care physician with 10/10 right knee pain and marked edema for 2 days. There was no erythema or warmth of skin. The patient denied fever and rigors. She underwent an MRI scan of the right knee on September 30, 2014. She developed a significant hemorrhagic effusion post procedure with purpura extending up towards the pelvis and down towards the feet. There was no apparent cause for the bleeding. She had heavy menses since menarche and underwent a hysterectomy for uncontrolled uterine bleeding secondary to fibroids and endometriosis. She reports several instances of post-operative bleeding such as post tonsillectomy and wisdom teeth extraction. Ms. Y reported that even as a toddler, she has been somewhat anemic. She has seen other hematologists in the past with no definitive diagnosis of bleeding disorder established. She also suffers from frequent migraines and complains of associated epistaxis. She has not used aspirin or NSAIDs since age 60.
Ms. Y’s past medical, family, and social history were insignificant for any type of bleeding disorder. She was not allergic to any medications. She was post-menopausal at age 39 and is estrogen dependent. She has no history of drinking, smoking, or contact with hazardous material. Ms. Y’s physical exam was insignificant without any hematologic abnormalities.
Ms. Y was worked up for bruising and bleeding that was postoperative and prolonged. Prothrombin time (PT) and partial thromboplastin time (aPTT) were normal. Factor VIII activity, factor IX activity, and von Willebrand panel are overall normal. Complements were normal as well. Factor XIII, α₂-antiplasmin, fibrinogen by clotting method, thrombin time, von Willebrand 2N were all within normal limits. However, the patient’s DNA was positive for PAI-1 Locus 4G/5G heterozygous for the 4G/5G deletion/ insertion allele. This was significant for intermediate PAI-1 activity and antigen levels compared to those individuals that have either the 4G/4G genotype with the highest PAI levels or 5G/5G genotype with the lowest PAI levels (Figure 2). Ms. Y was diagnosed with plasminogen activator inhibitor-1 deficiency.
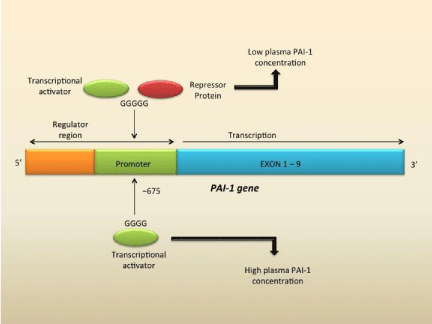
Figure 2: Structure of the gene for Plasminogen-Activator Inhibitor Type 1
(PAI-1) and the site of the 4G/5G polymorphism in the promoter region.
The 4G/5G polymorphism at position -675 influences transcription of PAI-1.
Increased transcription is associated with the 4G allele or four guanine bases
phenotype. This is because the 4G allele results in transcriptional activator
binding to the promoter. The 5G allele or five guanine bases causes the
binding of a repressor protein that decreases binding of the transcriptional
activator, leading to decreased PAI-1 plasma concentration. Ms. Y was
4G/5G heterozygote, thus with intermediate PAI-1 concentration, but still
manifested in the clinical symptoms of delayed postoperative bleeding. [2]
Figure adapted from [2] and created on Microsoft PowerPoint.
Conclusion
PAI-1 is an important down regulator of the fibrinolytic system [6]. Plasmin, the primary protease responsible for fibrinolysis, is formed from the cleavage of the zyomogen plasminogen. Two plasminogen activators, tissue-Plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA), catalyze the formation of plasmin. The efficiency of plasmin generation is increased by the fibrin clot, which provides a surface for formation of a ternary complex of fibrin, t-PA and plasminogen. Therefore, fibrinolysis exclusively occurs on the clot surface and not in circulation [6]. In order to control this process, enter plasminogen activator inhibitors, with the primary inhibitor being PAI-1. Part of the serpins, or superfamily of serine protease inhibitors, this 47-kDa protein binds to plasminogen activators with rapid and irreversible inhibition– suicide inhibition (Figure 1) [1,2,7,8]. PAI-1 is mainly produced by the endothelium, but can also be secreted by other tissue types such as adipose tissues. PAI-1 inhibits the serine proteases t-PA and u-PA, therefore inhibiting fibrinolysis [1].
To understand the end, one must start from the beginning. Ms. Y had an extensive history of bleeding including anemia, heavy menses, postsurgical bleeding after wisdom teeth, hysterectomy, and more recently, the hemarthroses. As a clinician, this type of delayed bleeding brings up a certain differential of bleeding disorders. Typically, by history, mucosal bleeding is caused by platelet quality/ quantity/functionality, vasculitis, or connective tissue disease. Ms. Y’s platelet function testing (PFA-100) was normal and her platelet numbers were sufficient. She had no obvious blood vessel issues or history and physical that pointed towards Osler-Weber-Rendu. Ms. Y’s history actually displays delayed type of bleeding, which is very similar to factor deficiencies.
Ms. Y had a normal aPTT, mildly low PT, and normal platelet count. Therefore, one must think of other reasons for this patient’s bleeding (Figure 3). Although not deficient, factor VIII and factor IX activity could be responsible, however their activities were normal. Typically, factor deficiencies such as factor VIII and IX (hemophilia) will have delayed bleed but often with PTT prolongation. These factors were normal. The panel for von Willebrand, to rule out von Willebrand deficiency, another common source of bleeding with normal PT/aPTT, was ordered and returned normal.
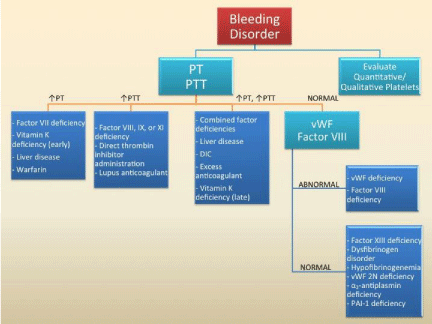
Figure 3: Diagnostic algorithm to approach bleeding disorders.
Prothrombin Time (PT); Partial thromboplastin time (PTT); Disseminated
Intravascular Coagulation (DIC); von Willebrand Factor (vWF); von Willebrand
Factor Type 2N or Normandy (vWF 2N); Plasminogen Activator Inhibitor Type
1 (PAI-1); Created on Microsoft Power Point.
At this point, Ms. Y had definitive delayed bleeding after surgery with no apparent etiology. The differential now includes more rare disorders including factor XIII deficiency, dysfibrinogen disorders, hypofibrinogenemia, von Willebrand disease type 2N (Normandy), α₂-antiplasmin deficiency and plasminogen activator inhibitor deficiency.
Factor XIII stabilizes and crosslink’s fibrin strands (Figure 1) [9]. Deficiency in Factor XIII can certainly be the cause in this patient as it typically causes delayed bleeding usually 24 to 36 hours after surgery or trauma [9]. Coagulation testing would have normal values for PT and aPTT. Factor XIII qualitative with reflex 1:1 mix was ordered. Clot lysis was not observed in the qualitative factor XIII screening test, suggestive of no factor XIII deficiency.
Fibrinogen is the substrate for fibrin clot formation, binds to platelets to support platelet aggregation, has a role in wound healing, and is a template for both thrombin and the fibrinolytic system (Figure 1) [10]. Due to her uncommon nature of disease, we suspected an abnormality in fibrinogen because most of the common causes have been ruled out. To measure, diluted plasma is clotted with a high concentration of thrombin. Clotting time of the dilution is established and measured against a sample of known fibrinogen concentrations [11]. Ms. Y was found to have normal fibrinogen levels.
Type 2N von Willebrand Disease (or Normandy, named after one of the first patients that were described) is an uncommon disorder that is inherited as an autosomal recessive trait [12]. Most mutations affect the N-terminus of the mature von Willebrand Factor monomer within the binding site for factor VIII, causing decreased binding to factor VIII. Unlike the other forms of von Willebrand’s disease, which have moderate to severe muco-cutaneous bleeding, Type 2N has a predilection to have delayed bleeding after invasive procedures, as seen in Ms. Y. However, Ms. Y tested negative to three of the common mutations: Thr791Met (15% incidence), Arg816Trp (10% incidence), and Arg854Gln (>70% incidence) [12].
Deficiency of α₂-antiplasmin is a rare congenital bleeding disorder resulting in enhanced plasmin activity and fibrinolysis (Figure 1) [13]. Since the blood coagulation system and platelets function normally in this type of deficiency, initial blood clotting after trauma is usually normal, with affected patient presenting with delayed bleeding as is our patient, Ms. Y. However, Ms. Y was tested for alpha-2 plasmin inhibitor and was within reference range.
Ms. Y’s DNA was evaluated for the PAI-1 4G/5G promoter polymorphism via polymerase chain reaction technology and restriction fragment length polymorphism. She was found to be heterozygous for the 4G/5G deletion/insertion allele. The 4G/5G genotype is associated with intermediate PAI-1 activity. 4G/4G genotypes have the highest PAI levels, whereas the 5G/5G genotypes have the lowest PAI levels [14,15]. Ms. Y was found in between (Figure 2).
PAI-1 has a similar picture for delayed bleeding seen in factor deficiencies. In hemostasis, clots are formed first, followed by fibrinolysis (Figure 1). Since PAI-1 deficiency affects fibrinolysis, it is easy to appreciate that the bleeding will be delayed. Ms. Y had multiple clinical histories of delay such as the bleeding into the knee, post dental work then bleed, and even heavy menses. Although these episodes are mucosal type bleeding, it is important to realize that these were delayed mucosal bleeds. For example, the menses clot was formed first, followed by bleed and the dental clot was formed first then followed by bleed. A true platelet dysfunction would have an immediate mucosal bleed not seen in Ms. Y.
PAI-1 deficiency is a rare bleeding disorder whose diagnosis is a challenge but not impossible. Although PAI-1 deficiency has genetic components, it is not typically detected in the young. As our patient, the first person diagnosed with PAI-1 deficiency was 76 years old [3]. Although complete deficiency can lead to severe hemorrhagic consequences [4], our patient had only intermediate decrease in PAI-1 activity, underscoring the importance of PAI-1 in the bodies’ fibrinolytic system. The idea is to consider PAI-1 deficiencies in patients with delayed post-traumatic or post-surgical bleeding after other more common bleeding disorders have been excluded. It is our hope that this case report will add to a future database of clinical manifestations of intermediate activity of PAI-1 deficiency, which can help define the wide spectrum of a disease not yet fully explained by medical text.
References
- Mehta R, Shapiro AD. Plasminogen activator inhibitor type 1 deficiency. Haemophilia. 2008; 14: 1255-1260.
- Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000; 342: 1792-1801.
- Schleef RR, Higgins DL, Pillemer F, Levitt LJ. Bleeding diathesis due to decreased functional activity of type 1 plasminogen activator inhibitor. J Clin Invest. 1989; 83: 1747-1752.
- Iwaki T, Tanaka A, Miyawaki Y, Suzuki A, Kobayashi T, Takamatsu J, et al. Life-threatening hemorrhage and prolonged wound healing are remarkable pehnotypes manifested by complete plasminogen activator inhibitor-1 deficiency in humans. J Thromb Haemost. 2011; 9: 1200-1206.
- Morimoto Y, Yoshioka A, Imai Y, Takahashi Y, Minowa H, Kirita T. Haemostatic management of intraoral bleeding in patietns with congenital deficiency of α2-plasmin inhibitor or plasminogen activator inhibitor-1. Haemophilia. 2004; 10: 669-674.
- Booth NA, Bachmann F. Plasminogen-plasmin system. Coleman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Philadelphia, PA: Lippincott Williams and Wilkins. 2006: 335-364.
- Huber K, Christ G, Wojta J, Gulba D. Plasminogen activator inhibitor type-1 in cardiovascular disease. Status report 2001. Thromb Res. 2001; 103: S7-19.
- Yepes M, Loskutoff DJ, Lawrence DA. Plasminogen activator inhibitor-1. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. Coleman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Philadelphia, PA: Lippincott Williams and Wilkins. 2006: 365-380.
- Kohler HP. Novel treatment for congenital FXIII deficiency. Blood. 2012; 119: 5060-5061.
- Collen D, Tytgat GN, Claeys H, Piessens R. Metabolism and distribution of fibrinogen. I. Fibrinogen turnover in physiological conditions in humans. Br J Haematol. 1972; 22: 681-700.
- Clauss A. [Rapid physiological coagulation method in determination of fibrinogen]. Acta Haematol. 1957; 17: 237-246.
- Meyer D, Fressinaud E, Gaucher C, Lavergne JM, Hilbert L, Ribba AS, et al. Gene defects in 150 unrelated French cases with type 2 von Willebrand disease: from the patient to the gene. INSERM Network on Molecular Abnormalities in von Willebrand Disease. Thromb Haemost. 1997; 78: 451-456.
- Saito H. α2-plasmin inhibitor and its deficiency states. J Lab Clin Med. 1988; 112: 671-678.
- Barcellona D. Thromb Haemost. 2003; 90: 1061. Dossenbach-Glaninger. Clin Chem. 2003; 49: 1081.
- Margaglione M, Cappucci G, Colaizzo D, Giuliani N, Vecchione G, Grandone E, et al. The PAI-1 gene locus 4G/5G polymorphism is associated with a family history of coronary artery disease. Arterioscl Thromb and Vasc Bio. 1998; 18: 152.