
Case Report
Ann Hematol Oncol. 2016; 3(1): 1071.
Secondary Autoimmune Myelofibrosis Associated with Rheumatoid Arthritis: A Rare but Steroid-Responsive Cause of Bone Marrow Failure
Reilly CR¹*, Sargent RL² and Sarkar M¹
¹Department of Medicine, Hospital of the University of Pennsylvania, USA
²Department of Pathology, Hospital of the University of Pennsylvania, USA
*Corresponding author: Reilly CR, Department of Medicine, Hospital of the University of Pennsylvania, 105 Wesley Avenue, Collingswood, NJ 08108, USA
Received: January 27, 2016; Accepted: March 07, 2016; Published: March 10, 2016
Abstract
Autoimmune myelofibrosis (AIMF) represents a distinct clinical and histopathologic entity characterized by progressive cytopenias due to bone marrow fibrosis in the context of a systemic autoimmune disease (secondary AIMF) or isolated autoantibody positivity (primary AIMF). Unlike primary myelofibrosis (PMF), AIMF typically exhibits a benign natural course and favorable response to corticosteroids. We present the case of a 35-yearold male with rheumatoid arthritis, who presented with fatigue, weight loss, dyspnea, diffuse lymphadenopathy, and progressive anemia and thrombocytopenia. Extensive workup did not reveal any systemic infection, hemolytic anemia, hypersplenism, malignancy, or other inflammatory/ autoimmune conditions. Bone marrow examination revealed hypercellular bone marrow with unremarkable trilineage hematopoiesis and diffuse, densereticulin fibrosis. There was no morphologic or immunophenotypic evidence of leukemia, lymphoma, myelodysplasia, or myeloproliferative neoplasm (MPN). Prednisone was initiated for the presumptive diagnosis of AIMF secondary to rheumatoid arthritis. Our patient demonstrated remarkable count recovery after six months of prednisone treatment with stable blood counts on low-dose maintenance steroids. While AIMF remains a rare diagnosis of exclusion, this entity should be considered in the differential diagnosis of patients with bone marrow fibrosis and autoimmune phenomena.
Keywords: Autoimmune myelofibrosis; AIMF; Rheumatoid arthritis
Case Presentation
A 35-year-old male presented to our hospital with generalized fatigue, night sweats, nonproductive cough, progressive dyspnea, and 11-kg unintentional weight loss over six months. The patient was diagnosed with severe seronegative rheumatoid arthritis at age 23; prior therapy included prednisone, hydroxychloroquine, methotrexate, mycophenolate mofetil, infliximab, and adalimumab at various times. Additional past medical history was notable for a sterile exudative pleural effusion that resolved spontaneously several years previously, avascular necrosis of his right humerus secondary to chronic steroids, and chronic Methicillin-resistant S. aureus (MRSA) osteomyelitis of his right femur, requiring multiple and prolonged courses of antibiotics. Due to infectious complications and lack of follow-up, the patient had not received immunosuppressive therapy for over 18 months prior to his presentation. The patient was not aware of any family history of autoimmune or hematological disease. He reported smoking cigarettes daily and denied alcohol or illicit drug use. Of note, the patient reported his rheumatoid arthritis had been well controlled off medications and denied arthralgias or significant joint stiffness.
His physical exam was notable for generalized cachexia, decreased breath sounds in the left lung fields, diffuse nontender lymphadenopathy (cervical, mandibular, supraclavicular, axillary, and inguinal lymph nodes), tender hepatomegaly, and Swan neck deformities and ulnar deviation of bilateral metacarpophalangeal joints; no active synovitis or splenomegaly was noted.
Initial laboratory data showed white blood count (WBC) of 6.9 x103 cells/μL with a normal differential, hemoglobin (Hgb) 9.5 mg/dL with a low reticulocyte index, and platelets of 54 x103 cells/μL. Blood work three months prior to admission was unremarkable except mild normocytic anemia (Hgb 11.2 mg/dL). Peripheral blood smear demonstrated thrombocytopenia, normocytic hypochromic anemia, and reactive lymphocytes; there was no evidence of platelet clumping, schistocytes, leukoerythroblastosis, or teardrop cells. Concurrent flow cytometry studies showed no evidence of acute leukemia or clonal lymphoproliferative disorder. Erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), lactate dehydrogenase (LDH), and serum IL-6 were elevated. Iron studies were consistent with mixed iron deficiency and inflammatory anemia. Rheumatoid factor and antinuclear antibodies (ANA) were both negative, but anti-cyclic citrullinated peptide (anti-CCP) was markedly elevated at 69 units. Direct antiglobulin test (DAT) was positive for IgG and C3 without hemolysis. Serum protein electrophoresis did not detect a paraprotein and quantitative immunoglobulins revealed isolated elevated IgG of 3340 mg/dL. Additional blood tests that were within normal limits or negative included liver function tests, basic metabolic panel, coagulation studies, serum B12 and folate, HIV antibodies, HHV8 polymerase chain reaction (PCR), EBV titers, IgG4 level, Beta-D glucan, Cryptococcus antigen, and Vitamin D 25-OH.
Computed Tomography (CT) of the chest, abdomen, and pelvis showed a moderate multiloculated left pleural effusion and diffuse lymphadenopathy involving bilateral cervical, axillary, mediastinal, retroperitoneal, and inguinal lymph nodes. Pleural fluid analysis revealed an exudative pleural effusion with normal adenosine deaminase level and negative stains, cultures, and cytology. Subsequent pleural biopsy and culture were also negative for infection or malignancy. Sequential left axillary and left cervical lymph node excisional biopsies demonstrated reactive follicular hyperplasia without evidence of infection, malignancy, or IgG4-related disease. Concurrent flow cytometry studies on both lymph node biopsies showed no evidence of a clonal lymphoproliferative disorder. Total body Positron Emission Tomography-Computed Tomography (PET-CT) scan exhibited multiple extensive FDG-avid mediastinal, hilar, axillary and retroperitoneal lymph nodes consistent with lymphoproliferative disease, autoimmune, or granulomatous disease.
Over several weeks, the patient’s cytopenias continued to worsen (nadir Hgb and platelets of 6.4 mg/dL and 14x103 cells/μL , respectively), eventually requiring intermittent transfusions. Bone marrow biopsy showed mild hypercellularity for age (80% overall) with unremarkable trilineage hematopoiesis (Figure 1), scattered lymphoid aggregates, and diffuse reticulin fibrosis compatible with myelofibrosis grade of MF-2 based on European Consensus Criteria (Figure 2). There was no morphologic, flow cytometric, or cytogenetic evidence of leukemia, lymphoma, myelodysplasia, or myeloproliferative neoplasm. Moreover, JAK2, MPL, and CALR mutational analyses were negative. The presumptive diagnosis of secondary autoimmune myelofibrosis (AIMF) was made, and the patient was started on 60mg of prednisone daily and discharged with hematology follow-up.
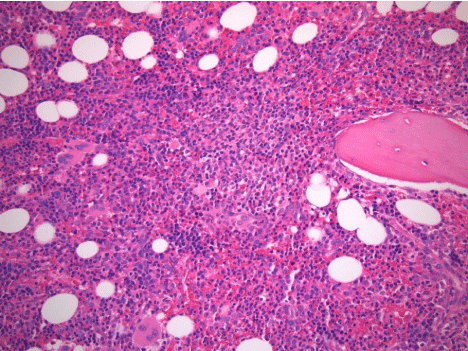
Figure 1: Core bone marrow biopsy with hypercellular marrow and trilineage
hematopoiesis (H&E, original magnification , 20x).
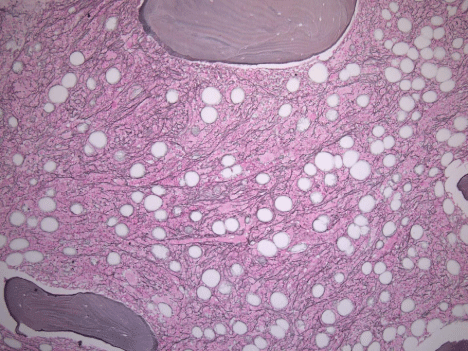
Figure 2: Diffuse and dense increase in reticulin fibers with numerous
intersections (Reticulin, original magnification, 10x).
After only one month of treatment, the patient’s thrombocytopenia and anemia improved markedly (Hgb 10.8 mg/dL and platelets 90x103 cells/μL) and the patient noted improved fatigue and dyspnea. After two months of treatment, the patient self-discontinued steroids for several weeks, which caused a precipitous drop in his platelet count that recovered quickly after resuming prednisone (Figure 2). Steroids were slowly tapered over six months and the patient is currently on low-dose maintenance prednisone and continues to have stable blood counts.
Discussion
AIMF represents a distinct clinical and histopathologic entity characterized by progressive cytopenias due to bone marrow fibrosis usually in the context of a systemic autoimmune disease [1-3]. Cavalcant first described the disease in 1978 in a young Chinese male with features of SLE, bone marrow fibrosis and refractory anemia that reversed with corticosteroid treatment [4]. In 1994, Paquette et al. described eight patients with SLE and myelofibrosis and coined the term “autoimmune myelofibrosis” [5]. Secondary AIMF has been observed in a wide array of other autoimmune conditions, including Sjogren’s syndrome, rheumatoid arthritis, autoimmune hemolytic anemia, antiphospholipid syndrome, Evan’s syndrome, autoimmune hepatitis, primary sclerosing cholangitis, Hashimoto’s thyroiditis, polymyositis, autoimmune demyelinating polyneuropathy, and pernicious anemia [3]. In time, primary AIMF was observed to occur in patients with autoantibody positivity in the absence of a clinical autoimmune disease [2]. Common autoantibodies seen in primary AIMF include ANA, RF, and DAT [2,3].
The diagnosis of AIMF is one of exclusion and requires systematic elimination of other etiologies of bone marrow failure, including MPNs, hematological malignancy, solid tumor metastases, infiltrative infections, medication side effects, and metabolic disorders [4]. AIMF typically exhibits a benign natural course and favorable response to corticosteroids [1]. In contrast, PMF has a dismal prognosis and inevitably progresses to transfusion-dependence and/or malignant transformation [4,6]. Given the significant variability in prognosis and treatment, it is imperative to distinguish AIMF from other more morbid bone marrow disorders.
The pathogenesis of AIMF is poorly understood, as is the mechanism of myelofibrosis in general. The putative mechanism involves activation and proliferation of marrow fibroblasts in response to cytokines released from megakaryocytes and platelets [7]. Activated fibroblasts produce excessive reticulin and collagen, which progressively effaces the marrow compartment producing peripheral cytopenias [7]. Several growth factors have been found to be necessary for marrow fibrosis, including TGF-B [8] and platelet derived growth factor (PDGF) [6]. Distinction is made in the literature between reticulin fibrosis and collagen fibrosis; the former occurs in a variety of benign conditions and demonstrates reversibility, whereas the latter is more indicative of a malignant process or MPN and often lacks reversibility [7]. Additionally, collagen fibrosis is associated with bone marrow osteosclerosis, peripheral leukoerythroblastosis, teardrop poikilocytosis, and splenomegaly, whereas patients with reticulin fibrosis typically lacks these features [7,9].
To date, there have been over 60 documented cases of primary and secondary AIMF in the literature [3,5]. The true incidence of AIMF is not known due to the rarity of the disease and lack of consistent diagnostic criteria. Among the reported cases of AIMF, there is considerable variability with respect to the presence of leukoerythroblastosis, teardrop cells, hemolysis, splenomegaly, bone marrow abnormalities (e.g. megakaryocyte dysplasia/clustering, osteosclerosis), and response to immunosuppression [1,3,5,10,11]. Of note, the majority of reported cases predate routine use of mutational analyses for MPNs, such as JAK2, MPL515, and CALR [3,8]. Therefore, it is probable that numerous reported cases of AIMF may in fact represent MPNs, which would potentially account for the lack of steroid-responsiveness in these patients [5].
Vergara-Lluri et al. detailed specific clinical and bone marrow findings that define primary and secondary AIMF in an attempt to standardize inclusion and exclusion criteria for diagnosis. In their cohort of 29 patients, the bone marrow of AIMF typically demonstrated hypercellularity (74%), reticulin fibrosis (100%), intrasinusoidal hematopoiesis (93%), lymphoid infiltrates and/or aggregates (83%), and the absence of atypical megakaryocytes, dysplasia, osteosclerosis, IgG4-positive plasma cells, MPNs, or malignancy [3]. In peripheral blood, AIMF does not exhibit significant leukoerythroblastosis or teardrop poikilocytosis characteristic of PMF or basophilia and eosinophilia typically of other MPNs. Cytopenias are nearly invariable at presentation with anemia and thrombocytopenia observed in 24% of cases. Intriguingly, rheumatoid arthritis was observed in 30% of the patients with secondary AIMF [3], whereas SLE was more commonly seen in other series [5].
Corticosteroids are the mainstay of treatment for AIMF; additional therapies used in the literature include azathioprine, mycophenolate mofetil, anti-thymocyte globulin (ATG), cyclosporine, danazol, colchicine, vincristine, intravenous immunoglobulin (IVIG), plasma exchange, and rituximab [3,10]. The majority of patients demonstrate improved cytopenias, and several patients have shown reversal of bone marrow fibrosis following treatment [3,5,10]. Six of seven patients with primary AIMF had a complete normalization of blood counts with a short course of prednisone (1mg/kg) for 1-3 months [2]. One case series of AIMF in SLE patients, 12 of 15 patients who underwent repeat bone marrow biopsy demonstrated improved reticulin fibrosis [10].
In conclusion, our patient presented with AIMF secondary to rheumatoid arthritis and demonstrated remarkable recovery of his blood counts that has persisted with tapering of immunosuppression. AIMF should be considered in the differential diagnosis of patients with bone marrow fibrosis and autoimmune phenomenon.
References
- Bass RD, Pullarkat V, Feinstein DI, Kaul A, Winberg CD, Brynes RK. Pathology of autoimmune myelofibrosis. A report of three cases and a review of the literature. Am J Clin Pathol. 2001; 116: 211-216.
- Pullarkat V, Bass RD, Gong JZ, Feinstein DI, Brynes RK. Primary autoimmune myelofibrosis: definition of a distinct clinicopathologic syndrome. Am J Hematol. 2003; 72: 8-12.
- Vergara-Lluri M, Piatek C, Pullarkat V, Siddiqi I, O’Connell C, Feinstein D, et al. Autoimmune myelofibrosis: an update on morphologic features in 29 cases and review of literature. Human Pathol. 2014; 45: 2183-2191.
- Cavalcant J, Shadduck RK, Winkelstein A, Zeigler Z, Mendelow H. Red-cell hypoplasia and increased bone marrow reticulin in systemic lupus erythematosus: Reversal with corticosteroid therapy. Am J Hematol. 1978; 5: 253-263.
- Paquette RL, Meshkinpour A, Rosen PJ. Autoimmune myelofibrosis. A steroid-responsive cause of bone marrow fibrosis associated with systemic lupus erythematosus. Medicine (Baltimore). 1994; 73:145-152.
- McCarthy DM. Annotation. Fibrosis of the bone marrow: content and causes. Br J Haematol. 1985; 59: 1-7.
- Kuter DJ, Bain B, Ghulam M, Bagg A, Hasserjian RP. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol. 2007; 139: 351-362.
- Martyre MC, Romquin N, Le Bousse-Kerdiles MC, Chevillard S, Benyahia B, Dupriez B et al. Transforming growth factor-beta and megakaryocytes in the pathogenesis of idiopathic myelofibrosis. Br J Haematol. 1994; 88: 9-16.
- Thiele J and Kvasnicka H.M. Grade of bone marrow fibrosis is associated with relevant hematological findings-a clinicopathological study on 865 patients with chronic idiopathic myelofibrosis. Ann Hematol. 2006; 85: 226-232.
- Chalayer E, Ffrench M, Cathebras P. Bone marrow fibrosis as a feature of systemic lupus erythematosus: a case report and literature review. Springerplus. 2014; 3: 349
- Santos F, Konoplev S, Lu H, Verstovsek S. Primary autoimmune myelofibrosis in a 36-year-old patient presenting with isolated extreme anemia. Leuk Res. 2010; 34: e35-37.