
Case Series
Ann Hematol Oncol. 2016; 3(1): 1074.
Acquired Hemophilia: A Report of 4 Cases
Karimi M1*, Abdolkarimi B1,2, Majid Naderi 3 and Parand S1
¹Hematology Research Center, Shiraz University of Medical Sciences, Iran
²Department of Pediatrics, Iran
³Children and Adolescents Health Research Center, Zahedan University of Medical Sciences, Iran
*Corresponding author: Mehran Karimi, Department of Pediatric Hematology-Oncology, Hematology Research Center, Shiraz University of Medical Sciences, Nemazee Hospital, Shiraz, Iran
Received: January 20, 2016; Accepted: March 08, 2016; Published: March 10, 2016
Abstract
Acquired hemophilia A is very rare and often causes life-threatening hemorrhage. We report 4 cases of this disorder in different settings. Case1 received activated protrombin concentrate complex (aPCC) and the result was excellent. Case 2 received IVIG and high dose factor ѴІІІ, prednisolon and azathioprin and long term follow-up was without bleeding. Case 3 was a postpartum acquired hemophilia A and received recombinant FVIIa, prednisolon, cyclophosphamide and she had complete response. Case 4 was a 74 year- old man diagnosed with renal cell carcinoma induced acquired coagulopathy. He died due to initial mismanagement with high dose plasma concentrated factor VIII. Acquired hemophilia A should be treated by combination of immunosuppressive agents, aPCC or rFVIIa. It should be taken into consideration that the degree of inhibitor levels has no direct relationship with severity of bleeding symptoms and high index of suspicion is necessary for early diagnosis.
Keywords: Acquired hemophilia A; Factor VIII inhibitor; Hemophilia; Immunosuppressive therapy
Introduction
Acquired hemophilia A (AHA) is very rare disease, especially in children. It is caused by production of antibody against coagulation factor VIII. It is associated with mortality and morbidity, so early diagnosis is very crucial.
In the 6th-7th decade of life, AHA is diagnosed in both sexes equally [1]. The final diagnosis was based on the time-dependent inhibitor of factor VIII in plasma and low plasma level of factor VIII
The isolated prolongation of activated partial thromboplastin time (APTT) after the addition of normal plasma is the primary screening test. This situation was mistaken with other acquired bleeding disorders such as disseminated intra vascular coagulation. More than 50% of cases are idiopathic, but the underlying conditions including auto immune disease, malignancy, infection, pregnancy, medications (especially antibiotics, psychiatric drugs) and immune modulating agents can cause AHA [2]. The mortality rate is estimated from 7.9% to 22%. Therefore, accurate and early diagnosis and treatment can prevent mortality and morbidity. We reported four cases with AHA who were presented with different clinical settings.
Case Presentation
Case 1
The patient was a 13 year-old boy who presented with right elbow (Figure 1) and right knee joint edema secondary to mild trauma in school. Sonography of right elbow and knee showed hemorrhage. Laboratory evaluations were done and showed mild anemia (HB=10.7 gr/Dl) and prolonged partial thrombin time (PTT=80 second).
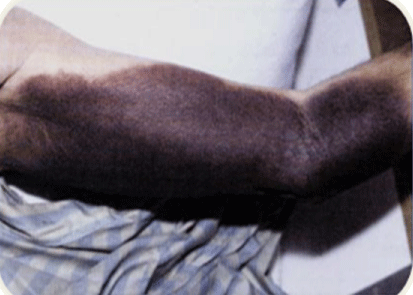
Figure 1: Right hemarthrosis in case 1 with acquired hemophilia A.
Coagulation assays showed prolonged mixing PTT, severe decreased factor VIII activity (< 1%) and inhibitor against factor VIII was 5.8 BU. Factor ІX, XI, XІІ & XΙΙΙ levels were normal. Also, prothrombin time, LE cell, Acla, Lac, ANA, Anti ds DNA, C3, C4, CH50, Liver Function Test (LFT), T3, T4, TSH were also normal
activated Prothrombin Complex Concentrated (aPCC, feiba) was given with dosage of 100 U/kg every 12 hours. The bleeding was stopped after 5 doses and result of treatment was excellent. The patient is in good condition at present with regular outpatient clinic follow-up.
Case 2
A 20 year- old woman presented with spontaneous left elbow hemorrhage without trauma (Figure 2). She had a mass 3x3 centimeters in left elbow. She had positive history of gum bleeding 6 months ago and received Fresh (Frozen Plasma (15 cc/kg) and Tranexamic acid with relative response.

Figure 2: Right knee hemarthrosis (left). Left elbow hemarthrosis (Right) in
patient case 2 acquired hemophilia A.
Laboratory evaluations showed normal complete blood count, PT, T4, TSH, and bleeding time but PTT was prolonged. Factor VIII activity was low (16.8%) and FVIII Inhibitor showed 2.3 BU.
ANA, Anti ds DNA Lac, ACla, Abdomen and pelvic sonography and CXR were normal. IVIG 1gr/kg for 2 days and high dose factor VІІІ (50 U/kg, every 8 hours) for one week followed by Azathioprin with initial dose of 2 mg/kg per day were started for this case. Inhibitor was negative and Factor VІІІ was elevated to 90% 2 months after treatment. Her follow- up during 1 year was good without any rebreeding episode.
Case 3
The patient was a 37 year- old woman. She had normal first vaginal delivery without history of coagulation disorders. Three months after second normal vaginal delivery she developed severe skin echymosis and bleeding of right upper extremity leading to compartment syndrome (Figure 3 A,B). Coagulation tests showed prolonged PTT and mixing PTT, low FVIII level (0.14%), and high FVIII inhibitor levels (145 BU). FIX, FX and FXII levels as well as PT and BT were normal.
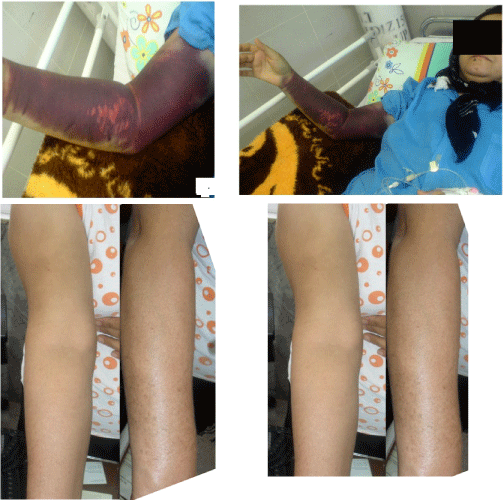
Figure 3: Diffused right upper extremity skin bleeding in a patient, case 3 with
acquired hemophilia A before (A, B) and after treatment (C,D) with rFVIIa,
prednisolon and cyclophosphamide (case 3).
ANA and, ds DNA, Acla and Lac were also normal. The patient was admitted in the central hospital and received plasmapheresis and recombinant FVII 90 u/kg (every 6 hrs) with poor response. Then the patient was referred to our tertiary hospital and the frequency of rFVIIa was changed to every 2 hrs for 24 hrs with complete response. The frequency was changed to every 4-6 hours for the second day. Immune suppressive treatment was started with prednisolone 1 mg/kg/d and cyclophosphamide 2 mg/kg/d at the same time. The coagulation tests showed normalization after completion of treatment. The bleeding symptom stopped after 2 days of starting acute bleeding episode. FVIII levels, inhibitor and PTT were 30%, 40 BU and 45 seconds respectively after treatment. The patient was discharged with continued prednisolone and cyclophosphamide for a period of 6 weeks with complete clinical response (Figure 3). She was followed for three years without any rebreeding episode.
Case 4
The patient was a 74 year-old man who presented with acute sudden extensive abdominal skin bruising and retroperitoneal bleeding. He was diagnosed as a renal cell carcinoma since 6 months earlier. FVIII levels and FVIII inhibitor were 8% and 5 BU respectively. The other coagulation tests were normal. The patient was diagnosed as AHA with no response to FVIII concentrate 50 U/kg every 8 hours and died due to refractory bleeding (Figure 4).
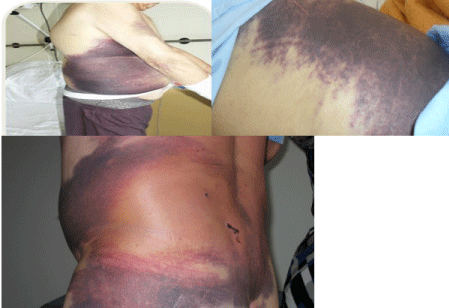
Figure 4: Diffused abdominal skin bleeding in right upper extremity in case 4.
Summary of coagulation tests in these 4 cases is shown in table one
![]()
Patient/TEST
Platelet
*PT
seconds
**PTT
seseconds nds
***BT
minute
Factor Ѵ III activity
VIII inhibitor
Treatment
outcome
Case 1
idiopathic
2211000
15
80
2
1%
3.8 B U
aPCC
Good
Case 2
idiopathic
330000
13.2
49.7
2:30
16.8%
1.3 BU
IVIG, n and high dose factor ѴІІІ, prednisolon and azathioprin
Good
Case 3
postpartum
26261000
13
55
102:10
0.14%
145 BU
recombinant FVII, prednisolon and cyclophosphamide
Good
Case 4
malignancy
351000
15
65
2.05
8%
5 BU
FVIIa concentrate
Poor
*PT::Prothrombin time
**PTT: Partial thromboplastin time
***BT: Bleeding time
Table 1: Coagulation Laboratory panel in four reported patients with AHA.
Discussion
AHA is a condition of potentially life-threatening bleeding situation caused by auto antibodies against FVIII. Acquired hemophilia A may be underestimated and given obscurity in making the diagnosis [4]. Auto antibodies have anti coagulating action mechanism and block the FVIII binding with Von Willebrand factor [5].
In case 1 and 2, autoimmunity was a new phenomenon which has short period of sustained period with other autoimmune disorders such as diabetes mellitus, Hashimoto thyroiditis, and vitiligo. Therefore, they require coincidental evaluation and outpatient followup. In approximately 50% of cases, there is an identifiable underlying clinical condition; in the other 50% no cause is known (idiopathic). In idiopathic AHA which resembles our case1, 2, coagulative supportive care is necessary for life saving intervention.
In contrast to congenital hemophilia, there is no correlation between bleeding symptoms and FVIII level or inhibitor [6]. Individuals with low-titer inhibitors can interact with the FVIII. The behavior of Auto-antibodies and congenital hemophilia A are different. Antibodies in congenital hemophilia have more FVIII neutralized activity than auto-antibodies in AHA, even the titer of auto antibodies in AHA may be higher than congenital hemophilia A [7].
In case 4 mismanagement occurred as a result of infusion of FVIII that was not appropriate for his management. Using inexpensive corticosteroid drugs with aPCC or FVIIa for this patient would have been life saving but the patient was diagnosed late with no response to FVIII concentrate, which resulted in his death. In this case, by passing agent like rFVIIa or a PCC was the first line of therapy. Due to severe bleeding symptoms, the low titer of inhibitor (5BU) and 8 % FVIII activity does not guarantee that our management concentrates would be effective. In fact, this case showed us that there is no direct concentration between factor VIII concentrate or inhibitor titer with bleeding symptoms in AHA
Normal history in case 3 showed rFVIIa with interval of every 6 hours cannot be effective unless it is changed to every 2-3 hours.
The development of high-titer inhibitors to FVIII and less often to other coagulation factors is the most serious complication of hemophilia therapy and makes treatment of bleeds very challenging. At present, by passing agents, such as Factor Eight Inhibitor Bypass Activity (FEIBA) and activated recombinant factor VII (rFVIIa) are the only coagulation factor concentrates available for the treatment of bleeds in inhibitor patients. Both products are effective and safe, and their efficacy has been found to be comparable (approximately 80%). A significant number of patients report a better effect of one or the other of the products, and in a minority of the patients none of the products are particularly effective. The haemostatic efficacy of bypassing agents is not considered equal to that of coagulation factor replacement in patients without inhibitors [8].
Auto antibodies do not bind the complement and do not induce allergic reactions. In more than half of the cases, the FVIII auto antibodies occurrence has no relation to any disorder or clinical condition (idiopathic AHA), and in these cases lupus anticoagulant does not just induce bleeding tendency, but also similar general population prone to thrombosis in up to 20% of cases. FVIII activity in healthy individuals range is among 50-150 % of normal values, and in AH patients is 0-15 % of normal values. AHA is more prevalent in older males, whereas young pregnant females may also be affected [5]. Postpartum AH usually happens in women within 3 months of delivery, most frequently after the initial delivery and usually does not reoccur with consequent pregnancies. Inhibitor may pass the placenta and continue for up to 3 months in the fetus, causing bleeding problem. It has been shown that rFVIIa is clinically safe and effective in the first and second line of treatments for severe bleeding episodes.
Diverse management modalities have been proposed for elimination of factor VIII inhibitor. Patients may respond to corticosteroid, cyclophosphamide, or combination of the current 2 drugs. In addition, high doses of intravenous gamma globulin have been administered but its effectiveness is questionable. In surgical procedures administration of aPCC and rFVIIa therapy for control of preoperative hemorrhage can be done safely in these patients. Combination therapy may be beneficial in control of bleeding episodes in AHA cases when mono therapy is ineffective, but it should be administered under caution and close monitoring [3].
AHA occurred in patients who had no history of previous bleeding disorder [9]. FVIII auto-antibodies can be acquired spontaneously in postpartum women. The diagnosis of post-partum AHA is highly based on clinical suspicion [6]. Theoretically, inhibitors develop more frequently during the postpartum period.
There is a hypothesis that mothers’ antibodies developed when exposed to fetal FVIII during the delivery. However, there was report of relapse on second pregnancies with postpartum in Kessler’s study in 1996 and FVIII inhibitors whose inhibitors had disappeared [10]. Prognosis is worse in patients with a high-inhibitor titer, low FVIII plasma activity, and high transfusional requirements. The previous observation revealed high-titer inhibitors (>10 BU) which do not easily disappear spontaneously and may be resistant to treatment with immunosuppressive therapy. On the other hand, if the inhibitor titer is low (<5 BU), this material generally disappears spontaneously and does not re occur on subsequent pregnancies. In case 3, we could stop bleeding by continuing bypassing agent (rFVIIa) for 2 days; otherwise, this compartment syndrome may have resulted in amputation (12). But in case 2, the patient had good response to IVIG and FVIII because she had low titer inhibitor.
Rituximab (anti- CD20 monoclonal antibody) and plasmapheresis were effective, especially in cases with very high-titer inhibitors [13]. Despite different mortality rates of 3.3 [11], thrombotic events were reported in 3.6% of patients treated with a haemostatic agent with a similar incidence between rFVIIa and aPCC [13]. In our cases, we did not find thrombotic event in using either rFVIIa or aPCC.
Factor VIII antibodies occurred in a close temporal relationship with the detection of many tumors. No association with a specific type of tumor could be identified. Immune suppressive treatment with prednisone +/- cyclophosphamide was successful in the majority of cases. However, azothropin was used instead of cyclophosphamide in case 2 because of the chance of infertility in young women using cyclophosphamide.
It is likely that there is a causal association between some solid tumors and factor VIII antibodies, but it is an extremely rare complication of cancer. The immunoglobulin nature of the inhibitor and the good response to immune suppressive treatment suggests that it is an autoimmune phenomenon. The pathogenesis is unknown. In case 4, if AHA had been considered by an expert hematologist and the patient had received appropriate treatment, he would have had a better outcome [15].
Conclusion
Acquired hemophilia is a very rare immune base condition, especially in children, that requires recognized underlying disorders and treatment with combination of immunomodulatory drugs and coagulative agents including FEIBA and rFVIIa which suppress antibody production against FVIII. AHA must be considered and highly suspicious whenever unexplained and sudden bleeding occurs in any case with negative history of previous bleeding. Coagulation work ups as well as finding underlying causes should be taken into account.
Acknowledgement
We thank Sheryl Nikpoor for editing and improving the use of English in the manuscript.
References
- Anna Buczma, Jerzy Windyga. Acquired haemophilia. Pol Arch Med. Wewn. 2007; 117: 241-245.
- Boţianu AM, Demian S, Macarie I, Georgescu D, Oltean G, Baţaga S. Acquired hemophilia complicated with gastrointestinal bleeding and spontaneous iliopsoas muscle hematoma in a woman with chronic C hepatitis under treatment with pegylated IFN alpha 2a and ribavirin. J Gastrointestin Liver Dis. 2012; 21: 93-95.
- Lak M, Sharifian RA, Karimi K, Mansouritorghabeh H. Acquired hemophilia A: clinical features, surgery and treatment of 34 cases and experience of using recombinant factor VIIa. Clin Appl Thromb Hemost. 2010; 16: 294-300.
- Shobeiri SA, West EC, Kahn MJ, Nolan TE. Postpartum acquired hemophilia (factor VIII inhibitors): a case report and review of the literature. Obstet Gynecol Surv. 2000; 55: 729-737.
- Scandella D, Mattingly M, de Graaf S, Fulcher CA. Localization of epitopes for human factor VIII inhibitor antibodies by immune blotting and antibody neutralization. Blood. 1989; 74: 1618-1626.
- Wootla B, Mahendra A, Dimitrov JD, Friboulet A, Borel-Derlon A, Rao DN, et al. Factor VIII-hydrolyzing IgG in acquired and congenital hemophilia FEBS letters. 2009; 583: 2565-2572.
- Campos-de-Magalhaes M, Eduardo Brandao-Mello C, Lucia Elias Pires M, Cecília da Fonseca Salgado M, Barcelo de Brito S, et al. Factor VIII and IX deficiencies related to acquired inhibitors in a patient with chronic hepatitis C virus infection receiving treatment with pegylated interferon plus ribavirin. Hematology. 2011; 16: 80-85.
- Tjønnfjord GE, Holme PA. Factor eight inhibitor bypass activity (FEIBA) in the management of bleeds in hemophilia patients with high-titer inhibitors. Vasc Health Risk Manag. 2007; 3: 527-531.
- Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Hemophilia. 2006; 12: 15-22.
- Cohen AJ, Kessler CM. Acquired inhibitors. Baillieres Clin Haematol. 1996; 9: 331-354.
- Hedner U, Erhardtsen E. Potential role for rFVIIa in transfusion medicine. Transfusion. 2002; 42: 114-124.
- Tagariello G, Sartori R, Radossi P, Gandini G, Franchini M. Intensive blood transfusion support in acquired hemophilia A. Ann Hematol. 2007; 86: 229-230.
- Onitilo AA, Skorupa A, Lal A, Ronish E, Mercier RJ, Islam R, et al. Rituximab in the treatment of acquired factor VIII inhibitors. Thromb Haemost. 2006; 96: 84-87.
- Baudo F, Collins P, Huth-Kuhne A, Levesque H, Marco P, Nemes L, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Hemophilia (EACH2) Registry. Blood. 2012; 120: 39-46.
- Hauser I, Lechner K. Solid tumors and factor VIII antibodies. Thromb Haemost. 1999; 82: 1005-1007.