
Case Report
Ann Hematol Oncol. 2016; 3(8): 1105.
Severe Secondary Hemophagocytic Lymphohistiocytosis Triggered by Chronic Active Epstein - Barr Virus Infection
Khoury J¹*, Alhalabi O¹, Haberichter K², Ogunleye F², Fennell T² and Jaiyesimi I³
¹Department of Internal Medicine, William Beaumont Hospital, USA
²Department of Pathology, William Beaumont Hospital, USA
³Department of Hematology and Oncology, William Beaumont Hospital, USA
*Corresponding author: John Khoury, Department of Internal Medicine, William Beaumont Hospital, 3601 W 13 Mile Rd, Royal Oak, MI 48073, USA
Received: June 13, 2016; Accepted: September 03, 2016; Published: September 06, 2016
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive and rare syndrome caused by pathologic activation of the immune system. It is classified into primary HLH, which is familial, and secondary HLH, which is acquired and associated with infections, malignancy and rheumatologic disorders. Here, we report a case of a 43-year-old male who presented with fever, elevated liver enzymes, hemolytic anemia and thrombocytopenia. The patient developed multiorgan failure and was transferred to the intensive care unit. Serological testing confirmed a previous Epstein–Barr virus (EBV) infection with positive EBV DNA in the blood detected by polymerase chain reaction (PCR). Further infectious workup from a nasopharyngeal swab was positive for rhinovirus detected by PCR. A diagnosis of HLH was established based on a bone marrow biopsy and the patient was started on induction therapy with intravenous dexamethasone and etoposide as part of the HLH-94 protocol. The patient gradually responded to therapy with full recovery after prolonged hospitalization
Keywords: Hemophagocytic lymphohistiocytosis; Chronic active EBV infection; HLH-94 protocol
Case Presentation
A 43-year-old Caucasian male who had splenectomy in his childhood due to idiopathic thrombocytopenic purpura (ITP) presented with jaundice and dark urine. The patient also reported flu like symptoms for a few days preceding his presentation to the hospital. He had no history of intravenous illicit drug use, alcohol use or liver disease. Shortly after being admitted, the patient became febrile (38°C) and tachycardic. He had scleral icterus with generalized jaundice and an unremarkable abdominal examination. Laboratory investigations revealed a white blood cell count of 30.0x103 cells/ mL (normal range: 3.5-10.1x103 cells/mL) with an absolute neutrophil count of 24.9x103 cells/mL (normal range: 1.6-7.2x103 cells/mL), hemoglobin of 11.8 g/dL (normal range: 13.5-17.0 g/dL), platelets 63x103 cells/mL (normal range: 150-400x103 cells/mL), alkaline phosphatase 77 U/L (normal range: 30-110 U/L), asparate aminotransferase (AST) 141 U/L (normal range: 10-37 U/L), alanine aminotransferase (ALT) 50 U/L (normal range: 9-47 U/L), total bilirubin 16.6 mg/dL (normal range: 0.3-1.2 mg/dL) with a direct bilirubin of 4.9 mg/dL (normal range: 0.0-0.3 mg/dL) at this point in time broad spectrum antibiotics were started for suspected sepsis. Other laboratory values included lactate dehydrogenase (LDH) 5238 U/L (normal range: 100-238 U/L), haptoglobin <8 mg/dL (normal range: 40-240 mg/dL), fibrinogen 412 mg/dL (normal range: 175-400 mg/dL) and a positive direct antiglobulin test (DAT). A peripheral blood smear did not show spherocytes, blasts or microangiopathic changes. Serologic studies were positive for EBV-IgG and nuclear antigen, while EBV-IgM and early antigen were negative. EBV DNA was detected in the blood by PCR. Other laboratory studies for viral and bacterial infections including blood cultures, hepatitis A, B and C, cytomegalovirus (CMV), mycoplasma, influenza A and B, HIV, adenovirus, parvovirus B19, human metapneumovirus, respiratory syncytial virus (RSV), and parainfluenza virus were negative. Nasopharyngeal swab polymerase chain reaction (PCR) testing for a broad panel of respiratory viruses was positive for rhinovirus. Other laboratory screening tests for antinuclear antibodies, rheumatoid factor, paroxysmal nocturnal hemoglobinuria and serum cryoglobulin were negative. On the third day of admission, the patient’s hemoglobin dropped to 4.1 g/dL. He was started on IV Dexamethasone and IVIG for presumptive autoimmune anemia and thrombocytopenia (Evans Syndrome). The patient was transferred to the intensive care unit for close monitoring. His platelets count improved, however despite multiple transfusions his hemoglobin continued to drop with a nadir of 3.0 g/dL. The patient continued to have persistent fever between 38°C and 39.5°C. He eventually developed multiorgan failure, which required mechanical ventilation and continuous renal replacement therapy (CRRT) support. To evaluate the underlying cause of the progressive anemia and thrombocytopenia the patient underwent a bone marrow biopsy which showed a hypercellular marrow with activated hemophagocytic macrophages (Figure 1). Flow cytometry on the bone marrow aspirate was negative for hematolymphoid malignancy. Further laboratory tests revealed a ferritin level of 62171 ng/dL (normal range: 14-338 ng/dL), triglycerides of 1310 mg/dL (normal range: 30-149 mg/dL) and a soluble CD25 (soluble IL-2 receptor alpha) level of 819 U/mL (normal range: 1033 U/mL or less). A diagnosis of HLH was made and subsequently the patient was started on IV Etoposide according to the HLH-94 protocol. MRI of the brain and CSF studies were negative for CNS involvement. Cyclosporine was not given due to impaired kidney function and the risk of developing posterior reversible encephalopathy syndrome (PRES). The patient also received Pneumocystis jiroveci pneumonia prophylaxis with trimethoprim-sulfamethoxazole throughout therapy. He responded gradually to therapy and was successfully extubated after 13 days of mechanical ventilation. Kidney and liver functions slowly recovered and his blood counts stabilized. He was discharged to the rehabilitation ward 52 days after admission. On the day of discharge his white blood cell count was 4.0x103 cells/mL, hemoglobin 8.1 g/dL, platelets 481x103 cells/mL, ferritin 3822 ng/ dL, triglyceride 169 mg/dL and liver function tests were normal. The patient has normal functional status 3 years after presentation. Follow up laboratory tests have shown normal blood counts in addition to normal kidney and liver function. Figure 2 illustrates the trend for pertinent laboratory values throughout the hospital stay.
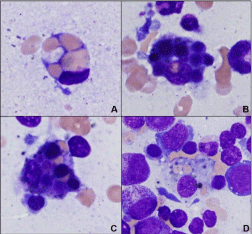
Figure 1: Bone marrow aspirate showing hemophagocytichistiocytes
with minimal nuclear atypia. This composite highlights the various
hemophagocytichistiocytes containing numerous ingested red blood cells
(image A), and small mature lymphocytes (image B). In addition, there
were histiocytes containing multiple mature hematopoietic cells including red
blood cells, lymphocytes and platelets (images C and D). (All images Wright
Giemsa stained, 1000x magnification).
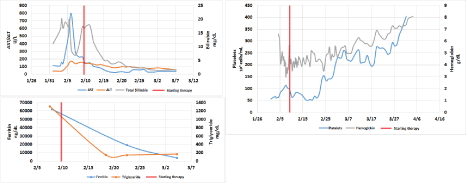
Figure 2: Trend in laboratory values subsequent to starting etoposide (VP-16) therapy (vertical red line).
Discussion
Hemophagocytic lymphohistiocytosis is classified into primary or inherited and secondary or acquired forms. Inherited forms are the main category in childhood HLH and have been attributed to genetic defects, including defective perforin and abnormal intracellular pathways required for the release of cytolytic granules by natural killer (NK) cells and cytotoxic T-lymphocytes [1]. Secondary HLH occurs most commonly in the setting of infections, malignancy, autoimmune disease or drug hypersensitivity reactions. Chronic active Epstein Barr virus (CAEBV) infection is a severe and often fatal disease with a complicated pathogenesis. It has a significant overlap with hemophagocytic lymphohistiocytosis in molecular targets within activated T-cell subpopulations, with defects in genes that are essential for regulating lymphocyte activation and proliferation. In both CAEBV and EBV-related HLH, T cells and NK cells are the main cell types infected [2]. It has been shown that EBV latent membrane protein-1 (LMP1)can activate cytotoxic T cells through decreased function of the signaling lymphocyte activation molecule (SLAM)– associated protein (SAP), resulting in the release of cytokines such as tumor necrosis factor alpha (TNF-α), interferon-gamma (IFN-γ) and granulocyte-macrophage colony stimulating factor (GM-CSF). These inflammatory cytokines in turn activate macrophages, resulting in delayed apoptosis, perforin deficiency, NK cell hypofunction, and ultimately persistent and uncontrolled hypercytokinemia. The sustained macrophage activation leads to unregulated inflammatory reaction characterized by extensive release of more cytokines (IL-1, IL-6), tissue infiltration and phagocytosis of autologous blood cell lines (Platelets, WBC and RBC) leading to multi-organ failure and other associated symptoms [3,4]. Most cases of CAEBV have been reported from Japan with rare cases in individuals other than Japanese children and adolescents [5]. High antibody titers against EBVrelated proteins are not necessary for the diagnosis, because there can be a lack of serologic response in patients with CAEBV infection. The presence of serological evidence of past EBV infection with the detection of viral DNA in the blood in addition to clinical presentation that meet HLH criteria is sufficient to make a diagnosis of chronic active EBV infection [6]. Even though our patient tested positive for rhinovirus, his presentation was thought to be mainly secondary to CAEBV given the known association between EBV and HLH and the overlapping pathogenesis between the two conditions. However, rhinovirus infection may have contributed to the development of HLH in our patient and cannot not be totally ruled out. According to the HLH-2004 trial, the diagnosis of HLH is established either by a molecular diagnosis consistent with HLH (primary HLH) or by the presence of five out of eight of the criteria listed in Table 1 [7,8]. Our patient presented with persistent fever, cytopenia of two cell lineages, hyperferritinemia, hypertriglyceridemia, and hemophagocytosis in the bone marrow aspirate, meeting the diagnostic criteria for HLH. The mortality of patients with hemophagocytic lymphohistiocytosis is very high without therapy. Retrospective reviews of the HLH-94 treatment protocol morbidity and mortality for 249 patients with longterm follow-up revealed a median survival of 54 percent at 6.2 years [9]. Therapy based on the HLH-94 protocol consists of eight weeks of induction therapy with etoposide (VP-16) and dexamethasone, with intrathecal Methotrexate therapy for those with CNS involvement [10]. The HLH-94 protocol also used cyclosporine starting at week nine. However, the use of cyclosporine is controversial as benefits are unproven as well as association with the development of posterior reversible encephalopathy syndrome (PRES).The major modifications in HLH-2004 protocol are the early use of cyclosporine during the induction phase of therapy and the addition of hydrocortisone to the intrathecal Methotrexate for patients with CNS involvement [7]. HLH-2004 study results have not yet been released; therefore, current recommendations are to initiate treatment based on the HLH- 94 protocol or on a clinical trial until the results of HLH-2004 are available [8]. Patients with HLH are at risk of developing infections during therapy, therefore prophylaxis for opportunistic organisms including Pneumocystis jirovecii and fungal organisms should be administered to all patients [8]. Hematopoietic cell transplant (HCT) is reserved for high risk patients including patients with HLH gene mutations, hematologic malignancy, relapsing symptoms, and central nervous system involvement [8]. Our patient received treatment with dexamethasone and etoposide with a very good clinical and laboratory response, and hematopoietic cell transplant was not necessary. He is still alive 3 years after presentation, despite his severe clinical condition, at least in part due to early diagnosis and treatment according to the currently recommended protocol.
![]()
Molecular identification of an HLH-associated gene mutation OR �At least five of the following eight findings:
- Fever =38.5°C
- Splenomegaly
- Cytopenias (affecting =2 of 3 lineages in the peripheral blood: hemoglobin <9 g/dL; platelets <100,000/microL; absolute neutrophil count <1000/microL
- Hypertriglyceridemia and/or hypofibrinogenemia (fasting triglycerides =3.0 mmol/L (i.e., =265 mg/dL), fibrinogen =1.5 g/L)
- Hemophagocytosis in bone marrow, spleen, lymph node, or liver
- Low or absent NK cell activity
- Ferritin >500 ng/mL
- Soluble CD25 (soluble IL-2 receptor alpha) =2400 U/ml
Table 1: HLH-2004 Diagnostic criteria [7,8].
References
- Janka G, ZurStadt U. Familial and acquired Hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2005; 82-88.
- Kasahara Y, Yachie A. Cell type specific infection of Epstein–Barr virus (EBV) in EBV-associated Hemophagocytic lymphohistiocytosis and chronic active EBV infection. Crit Rev Oncol Hematol. 2002; 44: 283-294.
- Aricò M, Danesino C, Pende D, Moretta L. Pathogenesis of haemophagocytic lymphohistiocytosis. Br J Haematol. 2001; 114: 761-769.
- Chuang HC, Lay JD, Hsieh WC, Wang HC, Chang Y, Chuang SE, et al. Epstein-Barr virus LMP1 inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome. Blood. 2005; 106: 3090-3096.
- Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, et al. Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United State. Blood. 2011. 117: 5835-5849.
- Okano M, Kawa K, Kimura H, Yachie A, Wakiguchi H, Maeda A, et al. Proposed Guidelines for Diagnosing Chronic Active Epstein-Barr Virus Infection. Am J Hematol. 2005; 80: 64-69.
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007; 48: 124-131.
- Jordan M, Allen C, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011; 118: 4041-4052.
- Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for Hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011; 118: 4577–4584.
- Henter JI, Aricò M, Egeler RM, Elinder G, Favara BE, Filipovich AH, et al. HLH-94: a treatment protocol for Hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. 1997; 28: 342-347.