
Review Article
Ann Hematol Oncol. 2016; 3(8): 1108.
Oncogenic Molecular Pathways: Mechanisms, Mutations and Inhibitors
Ali A1,2 and Li X1,2.*
¹Shanghai Key Laboratory of Regulatory Biology, East China Normal University, China
²Department of Molecular and Cellular Biology, Baylor College of Medicine, USA
*Corresponding author: Xiaotao Li, Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA
Received: July 26, 2016; Accepted: September 19, 2016; Published: September 21, 2016
Abstract
Oncogenic signaling pathways play crucial role in the development and progression of both solid tumors and hematological malignancies. It is well established that how these oncogenic signaling exerts its mode of action and how the key mutations in these signaling pathways lead to cancer development. Oncogenic signaling promote cancer progression by regulating growth, proliferation, cell cycle progression and apoptosis in both solid and hematological cancers. Activation of key oncogenic signaling is associated with drug resistance in multiple cancers, which is one of the main challenge to treat cancer patients effectively with chemotherapeutic agents. To date, several inhibitors are developed to target the key signaling molecules and mutations in these oncogenic signaling pathways to suppress tumorigenesis. Cell surface receptors and their cognate ligands play crucial role in the activation of cellular signaling, which subsequently enhance cancer metastasis and EMT-like features in cancer cells. Oncogenic signaling mainly exerts its mode of action via phosphorylation and activation of different transcription factors, and thereby control the expression of target gene, which are involve in tumorigenesis. Depletion or inhibition of key kinases in these oncogenic signaling pathways has been shown to suppress cancer cell growth and is associated with induction of apoptosis. Here, this review is focus on key oncogenic signaling pathways, their mutations and inhibitors in both solid tumors and hematological malignancies.
Keywords: Oncogenic signaling; Cancer progression; Chemotherapeutic agents; Growth factors; Genetic mutations; Leukemia
List of Oncogenic Molecular Pathways
- The Ras/Raf/MEK/ERK Pathway
- PI3K/ Akt Pathway
- PDGF signaling pathway
- JAK/STAT signaling pathway
- mTOR signaling
- NOTCH signaling
- Wnt signaling
- EGFR signaling
- MYC signaling
- Hedgehog (Hh) signaling pathway
Introduction
Tumor microenvironment has been shown to play crucial role in cancer cell growth, proliferation, DNA synthesis and survival and promotes epithelial mesenchymal transition (EMT)-like features and metastasis in both solid tumor and leukaemogenesis [1]. Several different types of inhibitors and cytotoxic drugs has been developed to suppress the growth and induce the apoptosis of cancer cells within tumor microenvironment. Genome instability, amplification, copy number alteration and mutations in key proteins play crucial role in cancer development, progression, EMT and metastasis [2]. Amplification of oncogenic proteins is key to tumorigenesis. Once, the amplification of oncogenic proteins occur, then its play a dominant role in cancer cells by suppressing the function of tumor suppressor genes and thereby promote cancer cell growth and proliferation. Protein-protein interaction play important role in the suppression function of tumor suppressor proteins. Oncogenic proteins mostly block the function of tumor suppressor proteins by interacting with key domain and region with tumor suppressor proteins and subsequently enhance cancer cell growth. Most of the oncogenic proteins serve as a transcription factors and they binds to the promoter regions of tumor suppressor genes and thereby repress their expression. In some cases, oncogenic proteins forms a large transcriptional complex, which also include co-activators and co-repressors, to suppress the function of tumor suppressor gene. In some cases, competition occurs among cancer-related proteins for binding to the promoters of their common target genes. In some solid tumors, oncogenic proteins co-amplification occurs, in which one of the protein play a dominant role to control cancer cell growth, proliferation and survival.
Both solid tumors and hematological malignancies show heterogeneity. The contain activation, amplification and mutations of several genes within the same tumor. Due to this heterogeneous nature of tumors, it is also challenging to suppress the cancer cell growth with different inhibitors, cytotoxic agents and monoclonal antibodies. Epigenetic changes has also been shown to play very important role in cancer progression and survival. Several studies linked chromatin modification, methylation and acetylation with cancer suppression or progression. The function of different coactivators and co-repressors in cancer cell in context-dependent.
The Ras/Raf/MEK/ERK pathway
Multiple growth factors proteins and cytokines binds to their target receptors, which is mainly express on cell surface, and triggers the activation of downstream Mitogen activated protein kinases (MAPK) that mainly include Ras/Raf/MEK/ERK signaling cascade [1]. This signaling cascade in turn activates and phosphorylates a network of transcription factors, which translocate into nucleus to control the expression of different genes that are involve in cell growth, invasion, migration and survival [2]. Several important proteins such as phosphatases, kinases and scaffold proteins play key role in the activation and amplification of MAPK regulated Ras/ Raf/MEK/ERK-dependent signaling pathway in cancer cells (Table 1). Importantly, genetic mutations and instability in key molecules of these signaling promote tumorigenesis, invasion and metastasis [3]. Mechanistically, Activation of MAPK occurs via association of a growth factor receptor (GFR) with Src homology 2 domain containing protein (Shc). Shc play crucial role in the recruitment of growth factor receptor-bound protein 2 (Grb2) proteins and the son of seven less (SOS) homolog protein to the cell surface receptor complex and thereby exchange occurs between GDP and GTP [4] (Figure 1). Moreover, it has been shown that insulin receptor substrate (IRS) also promote Ras signaling activation via binding with Grb2. To date, K-RAS, N-RAS and H-RAS, these three different types of Ras proteins exist. These three different proteins play important role in cancer cell in context-dependent manner [5]. It is well documented that GTPase activating proteins (GAPs) suppress the Ras signaling pathway. GAPs such as p120GAP and NF1promote the GTPase activity and thereby convert GTP to GDP to turn off Ras signaling pathway [6,7]. Once the Ras signaling becomes activated, then it activates and phosphorylate Raf protein to further activate the downstream signaling of MAPK. Ras mediated Raf-1 activation is dependent on calcium/calmodulin-dependent protein kinase II (CaMK-II), which phosphorylates Raf-1 at S338 [8]. B-RAF, RAF-1 (c-RAF) and A-RAF are the three different types of Raf, which exists to date. PP2A has been shown to play important role in inactivation of Raf-dependent signaling via dephosphorylation [9]. Furthermore, activated Raf phosporylate mitogen-activated protein kinase- 1/2(MEK1/2). In turn, activated MEK-dependent signaling pathway further phosphorylate and activates extracellular signal regulated kinases 1/2 (ERK1 and 2) by targeting T /Y residues [10] (Figure 1). Kinase suppressor of Ras (KSR) has been shown to suppress the MAPK-dependent signaling by targeting MEK via phosphorylation. Moreover, it is well established that p21-activating kinases (PAK) and Rac (Ras related gene) proteins also phosphorylate and activates MEK/ERK-dependent signaling in cancer cells. The dual specificity phosphatases DUSP (MKPs) act as negative regulator of MAPK signaling by inhibiting ERK1/2 signaling pathway in cancer cells [11]. Activated ERK1/2 signaling, in turn, phosphorylate and activate p90 Ribosomal six kinase-1 (p90Rsk1), MAPK signal integrating kinases (Mnk1/2), focal adhesion kinase (FAK) and myosin light polypeptide kinase (MLCK). Post-translational modification play important role in the activation of these target proteins by ERK1/2- dependent signaling [12]. ERK1/2-dependent signaling activates different transcription factors such as ETS, ATF-2, CREB and AP-1 to translocate into the nucleus. In nucleus, these transcription factors interact with different co-activators or co-repressors to regulate the target gene expression. These transcription factor mainly promote the expression of tumor associated genes and on another hand suppress the expression of tumor suppressor proteins to promote growth, invasion, proliferation, metastasis. Activated ERK1/2 signaling also has been shown to promote the expression of EMTrelated transcription factors such as TWIST1, SLUG, SNAIL and ZEB [13,14].
![]()
Inhibitors/agents
Target gene/protein/signaling
Cancer type
Clinical/preclinical /���� phase I, II and III trials
MAPK inhibitors
SB-590885
Raf
melanoma
Preclinical
BAY 43-9006
PDGFR, VEGFR
Leukemia, heptocellular carcinoma, melanoma
Phase I, II and III
PLX-4720
RAF
melanoma
Preclinical
RAF265
VEGFR,Raf
melanoma
Phase I
PLX-4032
Raf
Thyroid, melanoma, solid tumors
Phase I
CI-1040 (PD-184352)
MEK
Breast, lung, colorectal, pancreatic
Phase I, II
PD0325901
MEK
Breast, lung, colorectal, pancreatic, melanoma
Phase I, II
AZD6244
MEK
Liver, Breast, lung, colorectal, pancreatic, melanoma
Phase I, II
PD098059
MEK1/2
Leukemia, Liver, Breast, lung, colorectal, pancreatic
Preclinical
Tipifarnib
Ras
Leukemia, Breast, lymphoma
Phase I, II and III
U0126
MEK1/2
Leukemia, Breast, lymphoma, Liver, lung, colorectal, pancreatic
Preclinical
PDK/PI3K/AKT/mTOR inhibitors
BX-320
PDK
Leukemia, Breast, lymphoma, Liver, lung, colorectal, pancreatic
Preclinical
AR-12
AKT, PI3K
Breast, lymphoma, Liver, lung, colorectal
Phase I
KP372-1
AKT, PI3K
Brain, thyroid, leukemia
Preclinical
XL-147
AKT, PI3K
Lungs, breast, liver, colon
Phase I
GDC-0941
AKT, PI3K
Lungs, breast, liver, colon, lymphoma
Phase I
PX-866
AKT, PI3K
Lungs, breast, liver, colon, liver, pancreatic, ovary
Phase I
Celecoxib
PDK
Prostate, colon, lungs
Phase I and II
PWT-458
AKT, PI3K
Lung, brain
preclinical
Wortmannin
AKT, PI3K, mTOR, ERK, MEK
Brain, liver, lung, leukemia, breast, colon
preclinical
SR13668
AKT, PI3K
breast, colon, prostate, ovary
preclinical
MK-2206
AKT, PI3K
breast, lung, liver, colon, prostate, ovary, pancreatic
Phase I
CCI-779
mTOR
lung, leukemia, colon, breast
Phase I, II
AZD-8055
mTOR
breast, lung, liver, colon, prostate, ovary, pancreatic, lymphoma
Phase I, II
INK-128
mTOR
breast, lung, liver, colon, prostate, ovary, pancreatic, lymphoma
Phase I
OSI-027
mTOR
breast, lung, liver, colon, prostate, ovary, pancreatic, lymphoma
Phase I
WNT signaling inhibitors
OMP-54F28
WNT
breast, lung, liver, colon, prostate, ovary, pancreatic
Phase I
PRI-724
WNT/β-catenin
myeloma
Phase I
LGK974
WNT
Pancreatic, breast, melanoma
Phase I
Vantictumab
WNT
breast, lung, liver, colon, prostate, ovary, pancreatic
Phase I
CWP232291
WNT/β-catenin
leukemia
Phase I
Hedgehog signaling inhibitors
LDE-225
Hh
breast, lung, liver, colon, prostate, ovary, pancreatic, leukemia
Phase I, II
TAK-441
Hh
breast, lung, liver, colon, prostate, ovary, pancreatic
Phase I
LEQ-506
Hh
breast, lung, liver, colon, prostate, ovary, pancreatic
Phase I
GDC-0449
Hh
breast, lung, liver, colon, prostate, ovary, pancreatic, brain, myeloma
Phase II
BMS-833923
Hh
Gastric, breast, lung, liver, colon, prostate, ovary, pancreatic, brain
Phase I, II
JAK/STAT inhibitors
Pacritinib
JAK1/2/3
Leukemia, lymphoma
Phase I, II
Ruxolitinib
JAK1/2/3, TYK
Leukemia, breast, pancreatic
Phase II,III
AZD1480
JAK1/2/3
Gastric, breast, lung, liver, colon, prostate, ovary, pancreatic, brain
Phase I, II
SAR302503
JAK1/2/3, TYK
breast, lung, liver, colon, prostate
Phase I
Lestaurtinib
JAK2
Leukemia, lymphoma
Phase I, II
EGFR signaling inhibitors
ErlotinibHER1/EGFR
NSCLC
Phase II,III
Gefitinib
EGFR TKI
NSCLC
Phase II,III
Cetuximab
EGFR
NSCLC, head and neck
Phase II,III
Panitumumab
EGFR
Colorectal
Phase II,III
Sorafenib
EGFR, VEGFR
Heptocellular, renal
Phase II,III
sunitinib
EGFR TKI, VEGFR,PDGFR
gastrointestinal
Phase II,III
Lapatinib
HER2, EGFR
breast
PhaseIII
Table 1: Inhibitors.
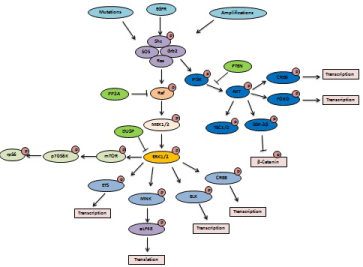
Figure 1: Activation of key oncogenic kinases signaling. Genetic mutations in receptors, amplification of oncogenes and growth factor receptors lead to
phophorylation of Ras, which in turn activates a signaling cascade. This oncogenic signaling cascade has cross talk with other signaling and results in the formation
of regulatory/feedback loops, which govern cancer progression and malignancy.
Ras/Raf/MEK/ERK pathway mutations: Mutations in the MAPK signaling pathways is crucial to cancer development and several mutations in this signaling pathway has been shown to promote cancer progression. Mutations in MAPK signaling pathway mainly over-express in tumors, where they also correlate with poor prognosis of cancer patients. About 20% of human cancers contain or have Ras mutations. Importantly, it has been shown that advanced pancreatic cancers patients contain about 70-80% mutations in K-Ras. Point mutations in RAS gene has been suggested to play important role in the activation of MAPK-dependent signaling and has been detected frequently in cancer patients. BRAF gene is frequently mutated in Langerhans cell histiocytosis (57%), melanomas (50-70%), and papillary thyroid cancers (PTC) (40%), about 2% in lung cancer and around 8% in colorectal cancers contains [15]. B-Raf inhibitors such as GDC-0879, dabrafenib and vemurafenib are mainly used to treat cancer patients that bear BRAF mutations. In addition, sorafenib, PLX-4720, and 885-A have ability to inhibit B-Raf mutants related cancer cell growth and proliferation. A MEK1/2 mutation has been found in lung and ovarian cancers [16]. U0126, PD98059, PD184352, PD0325901, RDEA119 and Selumetinib are used to treat cancer patients having activation of MEK-dependent signaling [17].
Ras/Raf/MEK/ERK pathway in leukemia: Ras/Raf/MEK/ERK signaling is also deregulated in leukemia and especially KRAS and NRAS mutations are mainly associated with Acute Lymphoblastic Leukemia (ALL). Ras pathway amplification is mainly caused by chromosomal translocation and somatic mutations in leukemia. FMS-related tyrosine kinase 3 (FLT3) mutations, BRAF mutations, protein tyrosine phosphates, non-receptor type 11 (PTPN11) somatic mutations, Neurofibromin (NF1) inactivation are mainly involved in activation of RTK signaling during leukemia development. About 2-9% cases of ALL involve FLT3 mutations. Importantly, FLT3 is activated in mixed-lineage-leukemia (MLL). PTPN11 is highly expressed by hematopoietic cells and mutations of PTPN11 occurs in 2-10% in ALL patients, while, about 35% of juvenile myelomonocytic leukemia patients represents PTPN11 mutations. BRAF mutations are very rare in ALL patients and predominantly amplification of BRAF involve in activation of Ras signaling during leukemia development. CBL mutations represent about 2% in Leukemia patients. Chromosomal translocation of BCR/ABL has been shown to involve in activation of Ras signaling in leukemia patients. Importantly, activation of Ras signaling show resistance to anti-cancer drugs in leukemia cells [18].
PI3K/AKT pathway
Phosphatidylinositol-3-kinase (PI3K) is a heterodimeric protein, which play important role in the migration, invasion and growth of cancer cells. Suppression of PI3K signaling has been linked with inhibition of tumor growth. PI3K contains a regulatory subunit and catalytic subunit (PIK3CA). PI3K have three different substrate specificity and lipid products: I, II, and III. PI3Ks Class I is further sub-divided into two type A and type B. Class IA PI3Ks has been shown to includes multiple regulatory subunits (p85-alpha, p85- beta, p55-alpha, p55- gamma, p50-alpha) and catalytic subunits (p110-alpha, p110-beta, p110- delta) [19], that play important role in PI3K-mediated signaling. PI3K class IB has been shown to composed of a p110-gamma catalytic subunit which binds the regulatory subunits, p101 and p87. PI3K play important role in the phosphorylation of membrane phospholipids signaling cascade, which include phosphatidylinositol 4-phosphate (PI4P) and PIP2 and PIP3 respectively [20]. PTEN is one of the most important negative regulator of PI3K signaling. PTEN exist tumor suppressor activity and its over-expression suppress the growth, migration and invasion of cancer cells. Furthermore, PH domain leucine-rich repeat protein phosphatase (PHLPP) has been shown to suppress AKTdependent signaling via dephosphorylation at S473 and thereby induces apoptosis in cancer cells [21].
PI3K/AKT pathway mutations: PI3K mutations have been linked with tumor progression of several cancers. Cancer patients having PI3K mutations show poor prognosis and poor survival rates. About 40% of ovarian cancers, 32% of colorectal, 30% of endometrial, 27% of brain, 25% of breast, 25% of gastric, 4% of lung cancers contain PI3K mutations [22]. Mutations of AKT1 are about 6% in colorectal cancer, around 8% in breast cancer, and 2% in ovary cancer. In contrast, AKT2 is not frequently mutated in human cancer and not play very aggressive role in cancer development. About 12.1% in ovarian cancer and 2.8% in breast cancer has been found to contain AK2 mutations [23]. Moreover, AKT3 mutations have been detected in some melanoma. Wortmannin, LY-294002, GDC-0941, PX-866, CAL- 101, XL-765 and XL-147 are used to inhibit PI3K pathway. OSU-03012 and Celecoxib are used to block PDK1 signaling. GSK690693, A- 443654, KP372-1, VQD-002, and Perifosine have ability to inhibit AKT signaling [24].
PI3K/AKT Pathway in leukemia: Activation of PI3K/AKT signaling play important in the cell growth, proliferation and development of leukemia. PDK1 activates AKT via phosphorylation by targeting threonine residue at 308 (Thr 308). About 50% of AML patients involve activation of AKT signaling at Thr308 residue. Moreover, AKT signaling activation via Thr308 is associated with poor survival of AML patients. Insulin-growth factor-1 signaling and FLTD3-IDT mutations has been shown to activate PI3K signaling during leukemia development (Figure 2) BCR-ABL oncogene mainly activates AKT signaling in B precursor lymphoblastic leukemi-ALL (Figure 4). It has been shown that p210 sub unit of BCR-ABL involve in interaction with PI3K via p85 subunit and hence activate PI3K signaling in Chronic Myelogenous Leukemia (CML). The p110d subunit of PI3K play crucial role in B cell signaling and has been shown the most important target for suppression of B-ALL. Deregulation of PI3K/AKT signaling has also been observed in leukemia initiating cells (LICs). Thus, PI3K/AKT signaling play important role in the progression of leukemia [25].
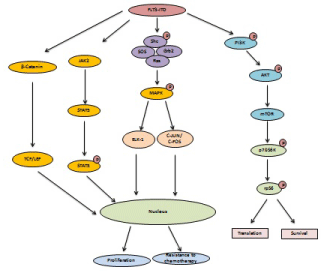
Figure 2: FLT3-ITD mutations involve in activation of MAPK, STAT signaling and β-Catenin signaling to promote drug resistance, cell survival and growth.
PDGF signaling pathway
Platelet-derived growth factor (PDGF) has been found to enhance the cell growth, motility, invasion and survival of cancer cells. Abnormal regulation of PDGF signaling is associated with the tumor development [26]. Over-expression of PDGF signaling has been linked with cancer development. Inhibition of PDGF suppresses cancer cell migration, invasion and growth. The PDGF mainly composed of composed of four different family members PDGF-AA, PDGF-BB, PDGF-CC and PDGF-DD, which exists in the form of homodimer [27]. PDGF receptor genes activation and mutations has been shown to promote cancer. The PDGFRa gene has point mutations in about 5% of gastrointestinal tumors [28]. Importantly, macrophages and endothelial cells play important role in PDGF secretion. PDGF secretion in turn activates PDGFdependent signaling pathway to control cancer cell growth and EMTlike features [29,30] (Figure 3). It has been shown that brain cancer cells and brain tumor tissues express high levels of PDGFR, which suggest the important and aggressive role of PDGFR in brain cancer development [31,32]. Moreover, PDGF receptors over-expression is also associated with prostate cancer development and angiogenesis. Depletion or inhibition of PDGFR is associated with suppression of growth and angiogenesis in prostate cancer. It has been also shown that over-expression of PDGFR promote liver cancer and its expression is negatively correlates with cancer patients survival [33]. Study on breast cancer cells also revealed that breast cancer cells express high levels of PDGFR, which play important role in migration and metastasis of breast cancer. Thus, targeting PDGFR receptor in different cancer is important for effective treatment of cancer patients. Sunitinib, imatinib, pazopanib, sorafenib, and nilotinib are the different anti-cancer drugs that has been shown to inhibit or block the PDGF receptor signaling in different cancers [34,35].
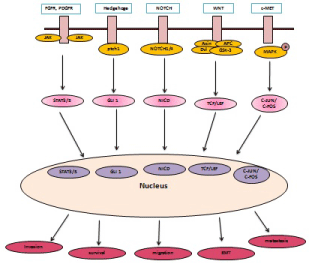
Figure 3: PDGFR, WNT, Hedgehog, NOTCH, c-MET signaling activates different transcription factors, which translocate to nucleus to promote cancer cell
invasion, migration, metastasis and EMT.
PDGF signaling in leukemia: It has been shown that PDGFRβ gene fusion occurs with TEL transcription factor in chronic monomyelocytic leukemia (CMML). Activation of PDGFRa has been associated with chronic eosinphilic leukemia. PDGFRa fusion with FIP1L1 (PDGFRa- FIP1L1) has been shown to activate STAT transcription factor family members including STAT1, STAT3 and STAT5 and MAPK family members such as p38 and ERK signaling eosinphilic leukemia. TEL-PDGFRa and TEL-PDGFRβ fusions has been shown to associated with resistance against chemotherapy such as imatinib. PDGF-BB in combination with IL-15 has been shown to promote large granular lymphocyte leukemia. Importantly, antibody targeting PDGF-BB has been shown to suppress the growth of leukemia cells. Thus, PDGFR signaling play important role in the development of leukemia [36].
JAK/STAT signaling
Abnormal regulation or over-expression of the JAK/STAT signaling pathway has been shown to promote cancer progression, invasion, migration and importantly inflammation [37]. JAK/STAT signaling pathway play critical role in the activation of inflammatory cytokines, that in turn promotes cancer metastasis. JAK family mainly consists of 4 different kinases named as JAK1, JAK2, JAK3, and TYK2. Different cytokines and inflammatory signaling such as IFN-γ, IL-20, IL-19 IL-6, IL-10, and IL-11 mainly activate JAK1 and JAK2. Whereas IL-3, IL-5, granulocyte macrophage colony-stimulating factor (GM-CSF), thrombopoietin and erythropoietin has been shown to enhance/activate the expression of JAK2 [38]. JAK2 activation has been shown to be important for recruitment of the STAT3 and STAT5 transcription factors to the receptor complex. JAK2-mediated STAT3 and STAT5 transcription factors phosphorylation results in the formation of homodimers and heterodimers complexes. Formation of these complexes is important for the translocation of STAT3 and STAT5 to nucleus, where they regulates the expression of target genes involved in cell growth, proliferation, cell survival and cancer progression [39]. Among different STAT family members, STAT3 and STAT5 has been shown to play very important and aggressive roles in the development of several cancers such as colon cancer, breast cancer, liver cancer and lung cancer [40]. LNK has been to act as negative regulator of JAK/STAT signaling, which inhibits these signaling to maintain a normal level of JAK/STAT proteins [41]. CYT387 has been shown to block JAK1/2 phosphorylation in cancer cells, which in turn leads to inactivation of STAT3 and STAT5 activation. Ruxolitinib has been shown to inhibit the phosphorylation of STAT5, ERK1/2, JAK1/2 and JAK2V617F in different cancer to suppress cancer cell growth. SAR302503 has been shown to suppress JAK2 and JAK2V617F. While, Pacritinib has been shown to specifically inhibit JAK2 signaling in cancer cells [42].
JAK/STAT signaling in leukemia: Deregulation of JAK/ SATAT signaling has been shown to play important role in the development of blood-related disease. A JAK1 mutation has been associated with adult lymphoblastic leukemia and poor prognosis in leukemia patients. Fusion of JAK2 gene with TEL (JAK2-TEL) has been found in B cell ALL and T cell ALL. JAK2 mutations fused with SSBP2 have been found in B cell ALL. STAT3 activation has been found in anaplastic large cell lymphoma and granular lymphocytic leukemia. STAT1 and STAT3 activation has been linked with chronic lymphocytic leukemia (CLL). About 20-50% AML patients show STAT3 activation. Importantly activation of STAT3 in AML is more common than STAT1 and STAT5, which also explain the important role of STAT3 activation in leukemia development [43].
mTOR signaling
The serine threonine kinase mammalian target of rapamycin (mTOR) plays an important and central role in protein translation and cancer cell invasion and migration [44]. Importantly, mTOR signaling has been shown to play critical role in cancer cell metabolism by regulating autophagy [44]. Over-expression of mTOR signaling components has been linked with cancer development and poor prognosis in cancer patients. Knockdown or suppression of mTOR components have been shown to suppress cancer cell growth and invasion. It is well established that Rapamycin and its analogs (rapalogs) are most effective anti-cancer drugs against mTOR signaling [45]. Rapamycin has been shown to suppress the cancer cell growth, invasion and migration that occurs due to over-expression of mTOR signaling. The mTOR signaling pathway mainly consists of mTOR complex 1 (mTORC1) and mTOR complexes 2 (mTORC2) [46]. Activation of mTORC1 subunit leads to phosphorylation of S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), which in turn promote translation. In contrast, activation of mTORC2 has been linked with activation and phosphorylation of PKC-dependent signaling pathway, AKT signaling and FOXO transcription factor, to promote cancer metastasis and EMT [47,48]. Several studies showed that modified rapamycins (rapalogs) such as CCI-779 (Temsirolimus, Torisel), rapamycin, RAD001 (Everolimus, Afinitor), AP-23573 (Ridaforolimus) are able to inhibit/suppress mTOR-dependent signaling pathway to overcome tumorigenesis in cancer cells [49].
mTOR signaling in leukemia: It has been shown that deregulated mTOR signaling promote drug resistance, show relapse and is associated with poor prognosis in AML patients. mTOR signaling play important role in the growth, proliferation and survival of leukemic cells. It has been shown that FLT3 activates mTOR signaling pathway and thereby maintains the survival of AML cells that involve FLT3 mutations. Spleen tyrosine kinase (SYK), T cell immunoglobin and mucin domain 3 (TIM3), C-KIT and Colony-stimulating growth factor 1 (CSF-1) have been shown to enhance mTOR signaling in AML cells. Importantly, interference with mTOR pathway via Raptor, which is the important subunit of mTORC1, has been shown to suppress the growth of leukemic cells in the bone marrow and blood of acute myeloid leukemia mouse model. Inhibition of mTOR signaling also has been linked with prolonged survival of AML mice. Thus, the role of mTOR signaling in progression and development of leukemia is well established [50].
NOTCH signaling
Aberrant regulation and activation of NOTCH signaling pathway and its different components has been shown to over-express in different cancers. NOTCH-dependent signaling play very aggressive role in various types of cancers and has been linked with cancer cell invasion, migration and proliferation (Figure 3). Inhibition of different NOTCH signaling components have been shown to suppress tumorigenesis [51,52]. NOTCH signaling mainly consist of four different NOTCH components that NOTCH1/2/3/4. These NOTCH family members have been shown to play critical role in multiple cancers that include breast cancer, liver cancer, colon cancer and lung cancer. Notch extracellular domain (NECD), which is composed of 29-36 repeated units of epidermal growth factor, has been shown to be important for proper folding of Notch receptors [53]. Moreover, NECD has been also shown to play important role in the interaction of NOTCH receptors with ligand Delta, Serrate, and Lag-2 (DSL) [54,55]. Tripeptide γ-secretase inhibitors such as IL-X (cbz-IL-CHO) and z-Leu-leu-Nle-CHO has been shown to inhibit/ suppress NOTCH-dependent signaling in various cancer cells to overcome tumorigenesis. Moreover, these inhibitors have also been shown to suppress the tumor growth in vivo that is mediated by NOTCH-dependent signaling. Several studies showed that MK0752 has ability to inhibit NOTCH-dependent signaling pathway that is over-expressed in advanced breast cancer patients [56].
NOTCH signaling in leukemia: About 60% of T-ALL patients show NOTCH signaling activation due to NOTCH 1 mutations. Thus, NOTCH1 mutation enhances NOTCH signaling in T-ALL leukemia to maintain the growth and survival of leukemic cells. Intriguingly, it has been shown that NOTCH signaling act as a tumor suppressive signaling in B-ALL, due to hypermethylation of NOTCH 3 and NOTCH signaling target genes Hes family members Hes2, Hes4 and Hes5 in B-ALL. In contrast, NOTCH signaling play oncogenic role B cell chronic lymphoblastic leukemia (B-CLL) by maintaining the survival, growth and proliferation of B-CLL. Targeting NOTCH signaling with gamma-secretase inhibitors (GSIs) has been shown to induce the cell death of B-CLL. NOTCH2 over-expression has been also implicated in B-CLL, which contributes to cell survival and suppresses apoptosis in B-CLL. It has been shown that activation of NOTCH signaling is associated suppression of chronic myeloid leukemia (CML) by inhibiting cell growth, proliferation and colony forming ability of K562 CML cells. Leukemic cells mainly dependent of the tumor associated microenvironment, which is known as leukemic microenvironment. Bone marrow stromal cells (BMSCs) play important role in the survival of leukemic cells. Several studies showed that NOTCH signaling is important for the cross talk of leukemic cells with BMSCs for their survival. Importantly, BMSCs has been shown to induce the expression of NOTCH1, NOTCH3 and NOTCH4, the key signaling molecules of NOTCH signaling. Thus, these results establishing the potential role of NOTCH signaling in leukemia and in BMSCs [57].
Wnt signaling
Wnt signaling pathways has been shown to play crucial role in embryonic development and has been linked with cancer progression [58]. Over expression of Wnt signaling pathway has been implicated in several cancers. In contrast, suppression or inhibition of Wnt signaling pathway has been show to suppress cancer cells growth, invasion and migration (Figure 3). It is well documented that Wnt signaling activity mainly dependent on β-catenin expression in the cytoplasm. Mechanistically, Wnt proteins binds to lipoprotein receptor-related protein/Frizzleds (LRP/Fz), which in turn phosphorylate a cytoplasmic dishevelled (Dvl) and thereby inhibits/ suppress the activity of GSK-3β activity. All these events, results in the accumulation of nonphosphorylated β-catenin in the cytoplasm [59]. β-catenin has been shown to recruit the transcriptional coactivators such as p300 and CBP to enhance the transcription of its target genes. Several studies showed that secreted Frizzled-related proteins (SFRP) and Wnt inhibitor factor 1 (WIF1) are important endogenous inhibitors of the Wnt signaling pathway [60,61]. Wnt/β- catenin signaling over-expression has been shown in several cancers that include breast cancer, liver cancer, lung cancer and colon cancer. Studies have indicated biological and small molecules inhibitors for the suppression of Wnt/β-catenin signaling. Importantly, studies have indicated that vitamin A and vitamin D3 have ability to inhibit Wnt/β-catenin signaling pathway in cancer cells [61]. Moreover, the inhibitory effect of polyphenols such as resveratrol, curcumin, epigallocatechin-3-gallate (EGCG) and quercetin on Wnt/β- catenin signaling pathway are also indicated by several studies [62]. Furthermore, small moleculae inhibitors such as FJ9, 3289-8625 and NSC668036, which target the PDZ domain in Dvl has been shown to inhibit Wnt-dependent signaling in cancer cells. In addition to this, several other inhibitors such as Inhibitors of Wnt response (IWR), Wnt production inhibitors (IWP) and XAV939 has been implicated in several studies to suppress/ inhibit Wnt/β-catenin pathway in cancer cells by targeting stabilization of Axin protein [63,64].
Wnt signaling in leukemia: Wnt signaling has been shown to over-express in AML and play important role in the self renewal of hematopoietic stem cells. Inhibition of Wnt signaling pathway has been linked with decrease proliferation in AML cell lines. Overexpression of Wnt signaling is associated with decrease survival of AML patients. Wnt signaling pathway proteins especially LEF1, Wnt1 and Wnt2B has been shown to over-express in AML patients. While, TCF/LEF transcription factors, which are activated by Wnt signaling pathway, has been shown to up-regulate in leukemic cells. Wnt signaling pathway also play important role in the development of leukemia stem cells. Moreover, Wnt signaling has also been shown to over-express in CLL and inhibition of Wnt signaling is linked with decrease cell survival and growth of CLL cell lines. Importantly, inhibition of wnt signaling in CLL cells is associated with increased apoptosis. Thus, these result suggest the oncogenic role of wnt signaling in the progression and development of leukemia [65].
EGFR signaling
It is noteworthy to mention that Epidermal growth factor receptor (EGFR) play critical role in cancer cell growth and has been shown to over-express in cancer patients. This signaling consist of four different family members that includes; EGFR (ErbB-1), HER2/c-neu (ErbB- 2), Her 3 (ErbB-3), and Her 4 (ErbB-4). Epidermal growth factors play central role in the activation of EGFR-dependent signaling in cancer cell. It has been shown that cancer cells over-express EGFR expression and suppression of EGFR activity has been linked inhibition of cancer cell growth. Activated EGFR signaling leads to activation of MAPK and PI3K signaling to enhance cell growth and DNA synthesis [66,67]. Several studies indicated that activated EGFR signaling pathway also plays a role in the suppression of autophagy in cancer cells [68]. EGFR signaling also has the ability to activate several transcription factors such as STAT3, STAT5, CREB and AP- 1, which play important role in regulation of inflammatory signaling. Cancer patients bears EGFR mutations are mainly treated with anticancer drugs such as gefitinib, erlotinib and afatinib [69,70].
MYC signaling
Role of MYC signaling in tumor progression is well established. MYC is a proto-ocogene and has been shown as a master transcription factor that has ability to control the expression of target genes involved in cell growth, cell proliferation and metastasis. MYC is over-expressed in various cancers and plays a crucial role in cancer development [71]. Inhibition of MYC activity has been linked with cancer suppression. Myc family proteins mainly consist of L-Myc, N-Myc, c-Myc, and Myc’s ‘second-cousins’ S-Myc and B-Myc [72]. Studies have shown that C-MYC is the most important for cancer cell growth and DNA synthesis and play aggressive role in tumor formation. It is well established that activation of MYC leads to phosphorylation of MAPK, AMPK and PI3K signaling in cancer cells to support the cancer cell survival. In contrast, Receptor tyrosine kinases and various growth factor receptors such as EGF, FGF, HGF has been shown to induce Myc expression in cancer cells. Importantly, studies have indicated that MLN8237/alisertib, which is an inhibitor of aurora A kinase, has the ability to suppress/inhibit MYC over-expression in tumors in clinical trials [73].
MYC signaling in leukemia: MYC signaling play important role in the progression of leukemia and has been shown to be upregulate in ALL via multiple mechanisms. About 2-5% children ALL and 5% adult ALL patients represents deregulation of MYC signaling. Lymphoblastic leukemia cells show high expression of MYC protein. Moreover, NOTCH signaling and TEL2 play important role in the induction of MYC expression in leukemia. Importantly, CLL represents decrease expression of MYC. AML patients show overexpression of MYC and high expression of MYC is important for the survival and proliferation of AML cells. The fusion of different transcription factors that involve in the progression of leukemia such as PLZF-RARa has been shown to activate the MYC gene expression to support AML cell survival. High expression of MYC is also reported in CML. It has been shown that BCR-ABL kinase induces MYC expression in CML to promote CML cell growth and proliferation (Figure 4) MYC over-expression is also linked with abnormal DNA synthesis in CML cells, which indicate the oncogenic role of MYC in CML patients [74].
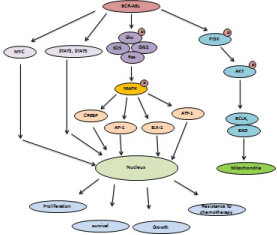
Figure 4: BCR-ABL fusion is involved in activation of MYC, MAPK, PI3K/AKT and STAT signaling to enhance cancer progression.
Hedgehog (Hh) signaling pathway
Hedgehog (Hh) signaling pathway was first reported in Drosophila and has been shown to play a potential role in proliferation, cell migration and differentiation [75] (Figure 3). Sonic Hedgehog (SHh), Desert Hedgehog (DHh) and Indian Hedgehog (IHh) are the family members of Hh signaling pathway. Hedgehog (Hh) signaling pathway is activated when Sonic Hedgehog (SHh), Desert Hedgehog (DHh) or Indian Hedgehog (IHh) binds to Patched (Ptch1) [76] . Upon binding of Hh ligand to Ptch, the expression of Gli transcription factor is changed and subsequently Smo is released. Gli is a zinc-finger transcription factor and has three types known as Gli1, Gli2 and Gli3 [77,78]. Hh signaling over-expression has been implicated in a variety of cancers. Studies indicated that Hh signaling components play important role in tumor progression. Aberrant regulation of Hh signaling components has been linked increased DNA synthesis, cell migration, cell invasion, inhibition of apoptosis. Activation of Hh signaling pathways is also associated with drug resistance in cancer cells. GDC-0449 and BMS-833923 (XL139) has been shown to block Hh pathway during cancer progression. GANT-58 and GANT- 61 are GLI antagonists and has the ability to suppress/inhibit deregulated Hh pathway in cancer cells. Importantly, Hh pathway inhibitor (HPI) such as HPI-1/2/3/4 has been shown to GLI transcription factor activity and subsequently suppress Hh dependent signaling pathway in cancer cells. In addition, studies have shown that Robotnikinin has ability to inhibit sonic Hh signaling [79-81].
Hedgehog (Hh) signaling in leukemia: It has been shown that Hh signaling is required for the expansion of leukemia stem cells. Moreover, Hh signaling also play important role in the growth, proliferation, survival and maintenance of leukemic cells. Importantly, Hh signaling has been shown to over-express in AML patients and AML cell lines. GLI-1 and SHH, the components of Hh signaling, has been shown to highly express in different leukemic cells including AML cell lines. High expression of GLI-1 is associatedv with decrease survival and drug resistance in AML patients. High expression of Hh signaling components also reported in B-CLL. High expressions of Hh signaling promote the expression of BCL-2 and thereby inhibit the apoptosis of leukemic cells and promote cell survival. Thus, Hh signaling pathway components play critical roles in the leukemia progression, which also make Hh signaling is one of the most important target in leukemia disease [82,83].
Conclusion
In this review, we have discussed various oncogenic pathways and kinases, which lead to cancer development and involve in the leukemia progression. We also discussed genetic mutations in the key proteins within these signaling pathways that support cell survival, cell growth and proliferation. Mutations in these proteins lead to abnormal regulation of oncogenic signaling and cellular transformation. In vivo studies indicated that over-expression of oncogenic signaling is associated with tumor formation ability. Genetic mutations and amplification of these proteins show resistance to anticancer agents, inhibitors and monoclonal antibodies, and most of the targeted therapy is short-time and less effective. Importantly, several studies showed that some of the key oncogenic signaling proteins are involved in the relapse and recurrence. Therefore, it is challenging to get rid off from the tumors completely due to amplification of these oncogenic signaling. Another important reason is that most of the tumors are heterogeneous in nature and contain more than one type of mutations in single tumor. Currently, combinations of different inhibitors are under clinical trials to improve and prolong cancer patients’ survival. Amplification of these oncogenic proteins also negatively correlates with survival of cancer patients. Importantly, researchers are focus on the development of specific inhibitors, small molecules, peptides, monoclonal antibodies and anti-cancer agents to target specific type of cancer. In case, if, the tumor growth depends on MAPK signaling pathway, then it will show sensitivity to MAPK inhibitors, so called “personalized therapy”. Recently, some clinical trials suggest that dual inhibitors show much better effect than single inhibitor alone due to heterogeneity. Importantly, these oncogenic signaling forms positive and negative feedback loops with each other during cancer progression, which is important for cell survival. Cancer microenvironment also play important role in the resistance of cancer cells against inhibitors, peptides, monoclonal antibodies and cytotoxic agents. Tumor microenvironments secrete different growth factors and cytokines, which promote inflammatory singling in cancer cells. Oncogenic signaling pathways such as STAT3 and MAPK have been shown to promote inflammatory cytokines production. Thus, it’s challenging to inhibit, block or suppress one specific targeted oncogenic signaling effectively in clinical trials and in cancer cells due to complicated links among these signaling.
Acknowledgement
This work was supported by the National Basic Research Program (2011CB504200) and in part by the National Natural Science Foundation of China (81261120555, 30870503, 81071657, 31100946); the Science and Technology Commission of Shanghai Municipality (11DZ2260300, 11ZR1410000). This manuscript was also funded by National Institutes of Health (1R01CA131914 and HD08818), Norman Hackerman Advanced Research Program (1082318401; PN004949-0012-2009).
References
- Steelman LS, Chappell WH, Abrams SL, Kempf CR, Long J, Laidler P, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011; 3: 192-222.
- McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F, et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011; 226: 2762-2781.
- Hafsi S, Pezzino FM, Candido S, Ligresti G, Spandidos DA, Soua Z, et al. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance. Int J Oncol. 2012, 40: 639-644.
- McKay MM, Ritt DA, Morrison DK. RAF inhibitor-induced KSR1/B-RAF binding and its effects on ERK cascade signaling. Curr Biol. 2011; 21: 563-568.
- McKay MM, Freeman AK, Morrison DK. Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases. 2011; 2: 276-281.
- Eblen ST, Slack JK, Weber MJ, Catling AD. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol 2002; 22: 6023- 6033.
- Balan V, Leicht DT, Zhu J, Balan K, Kaplun A, Singh-Gupta V, et al. Identification of novel in vivo Raf-1 phosphorylation sites mediating positive feedback Raf-1 regulation by extracellular signal-regulated kinase. Mol Biol Cell. 2006; 17: 1141- 1153.
- Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005; 17: 215-224.
- Sturm OE, Orton R, Grindlay J, Birtwistle M, Vyshemirsky V, Gilbert D, et al. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci Signal. 2010; 3: ra90.
- Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O, et al. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009; 16: 294-303.
- Buday L, Warne PH, Downward J. Downregulation of the Ras activation pathway by MAP kinase phosphorylation of Sos. Oncogene. 1995; 11: 1327-1331.
- Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ, et al. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998; 281: 1668-1671.
- Ekerot M, Stavridis MP, Delavaine L, Mitchell MP, Staples C, Owens DM, et al. Negative-feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008; 412: 287-298.
- Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004; 68: 320-344.
- Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: Rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011; 2: 135-164.
- Zheng Y, Xia Y, Hawke D, Halle M, Tremblay ML, Gao X, et al. FAK Phosphorylation by ERK primes Ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell. 2009; 35: 11-25.
- Steelman LS, Abrams SL, Shelton JG, Chappell WH, Bäsecke J, Stivala F, et al. Dominant roles of the Raf/MEK/ERK pathway in cell cycle progression, prevention of apoptosis and sensitivity to chemotherapeutic drugs. Cell Cycle. 2010; 9: 1629-1638.
- Knight T, Elizabeth Irving JA. Ras/Raf/MEK/ERK Pathway Activation in Childhood Acute Lymphoblastic Leukemia and Its Therapeutic Targeting. Front Oncol. 2014; 4: 160.
- Shull AY, Latham-Schwark A, Ramasamy P, Leskoske K, Oroian D, Birtwistle MR, et al. Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One. 2012; 7: e43369.
- Martelli AM, Evangelisti C, Chappell W, Abrams SL, Bäsecke J, Stivala F, et al. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011; 25: 1064-1079.
- Adams JR, Schachter NF, Liu JC, Zacksenhaus E, Egan SE. Elevated PI3K signaling drives multiple breast cancer subtypes. Oncotarget. 2011; 2: 435-447.
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3- phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997; 7: 261-269.
- Anderson DH. p85 plays a critical role in controlling flux through the PI3K/PTEN signaling axis through dual regulation of both p110 (PI3K) and PTEN. Cell Cycle. 2010; 9: 2055-2056.
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Ann Rev Pathol Mech Dis. 2009; 4: 127-150.
- Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015; 3: 2.
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999; 79:1283-1316.
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22: 1276-1312.
- Heldin CH. Targeting the PDGF signaling pathway in the treatment of non-malignant diseases. J Neuroimmune Pharmacol. 2014; 9: 69-79.
- Fredriksson L, Li H, Fieber C Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004; 23: 3793-3802.
- Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004; 15: 197-204.
- Ustach CV, Kim HR. Platelet-derived growth factor D is activated by urokinase plasminogen activator in prostate carcinoma cells. Mol Cell Biol. 2005; 25: 6279-6288.
- Ustach CV, Huang W, Conley-LaComb MK, Lin CY, Che M, Abrams J, et al. A novel signaling axis of matriptase/PDGF-D/β-PDGFR in human prostate cancer. Cancer Res. 2010; 70: 9631-9640.
- Omura T, Heldin C-H, Östman A. Immunoglobulin-like domain 4-mediated receptor-receptor interactions contribute to plateletderived growth factor-induced receptor dimerization. J Biol Chem. 1997; 272: 12676-12682.
- Yang Y, Yuzawa S, Schlessinger J. Contacts between membrane proximal regions of the PDGF receptor ectodomain are required for receptor activation but not for receptor dimerization. Proc Natl Acad Sci USA. 2008; 105: 7681-7686.
- Baxter RM, Secrist JP, Vaillancourt RR, Kazlauskas A. Full activation of the platelet-derived growth factor beta-receptor kinase involves multiple events. J Biol Chem. 1998; 273: 17050-17055.
- Chiara F1, Bishayee S, Heldin CH, Demoulin JB. Autoinhibition of the platelet-derived growth factor β receptor tyrosine kinase by its Cterminal tail. J Biol Chem. 2004; 279: 19732-19738.
- Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998; 93: 385-9539.
- Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007; 7: 673-683.
- Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000; 95: 19-29.
- Xiang Z, Zhao Y, Mitaksov V, Fremont DH, Kasai Y, Molitoris A, et al. Identification of somatic JAK1 mutations in patients with acute myeloid leukemia. Blood. 2008; 111: 4809-4812.
- Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365: 1054-1061.
- Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010; 115: 3109-3117.
- Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004; 117: 1281-1283.
- Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013; 32: 2601-2613.
- Sato T, Nakashima A, Guo L, Tamanoi F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem. 2009; 284: 12783-12791.
- Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Menendez JA. Raptor, a positive regulatory subunit of mTOR complex 1, is a novel phosphoprotein of the rDNA transcription machinery in nucleoli and chromosomal nucleolus organizer regions (NORs). Cell Cycle. 2011; 10: 3140-3152.
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009; 137: 873-886.
- Green AS, Chapuis N, Lacombe C, Mayeux P, Bouscary D, Tamburini J. LKB1/AMPK/mTOR signaling pathway in hematological malignancies: from metabolism to cancer cell biology. Cell Cycle. 2011; 10: 2115- 2120.
- Jiang Y. mTOR goes to the nucleus. Cell Cycle. 2010; 9: 868.
- Carew JS, Kelly KR, Nawrocki ST. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol. 2011; 6: 17- 27.
- Carneiro BA, Kaplan JB, Altman JK, Giles FJ, Platanias LC. Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol Ther. 2015; 16: 648-656.
- Wang Z, Li Y, Sarkar FH. Notch signaling proteins: legitimate targets for cancer therapy. Curr Protein Pept Sci. 2010; 11: 398-408.
- Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008; 111: 2220-2229.
- Gallahan D, Callahan R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4). Oncogene. 1997; 14: 1883-1890.
- Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004; 14: 779-786.
- Stylianou S, Clarke RB, Brennan K. Aberrant activation of Notch signaling in human breast cancer. Cancer Res. 2006; 66: 1517-1525.
- Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Hagenbeek Therapeutic antibody targeting of individual Notch receptors. Nature. 2010; 464: 1052–1057.
- Liu N, Zhang J, Ji C. The emerging roles of Notch signaling in leukemia and stem cells. Biomark Res. 2013; 1: 23.
- Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006; 5: 997-1014.
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997; 11: 3286-305.
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127: 469-480.
- van Amerongen R, Mikels A, Nusse R. Alternative wnt signaling is initiated by distinct receptors. Sci Signal. 2008; 1: re9.
- Wang Y. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther. 2009; 8: 2103-2109.
- Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009; 10: 468-477.
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007; 17: 45-51.
- Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJ. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia. 2012; 26: 414–421.
- Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: Egfr gene and cancer. FEBS J. 2010; 277: 301-308.
- Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: From development to tumorigenesis. Differentiation. 2007; 75: 770-787.
- Schneider MR, Wolf E. The epidermal growth factor receptor ligands at a glance. J cell physiol. 2009; 218: 460- 466.
- Gazdar AF. Epidermal growth factor receptor inhibition in lung cancer: The evolving role of individualized therapy. Cancer metastasis rev. 2010; 29: 37-48.
- Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R. Epidermal growth factor receptor - mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res. 2006; 12: 7261-7270.
- Soucek L, Whitfield JR, Sodir NM, Massó-Vallés D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes & development. 2013; 27: 504-513.
- Allen TD, Zhu CQ, Jones KD, Yanagawa N, Tsao MS, Bishop JM. Interaction between MYC and MCL1 in the genesis and outcome of non-small-cell lung cancer. Cancer research. 2011; 71: 2212-2221.
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146: 904-917.
- Chi Van Dang. Myc Roles in Hematopoiesis and Leukemia. Genes Cancer. 2010; 1: 605–616.
- Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009; 9: 873-886.
- Yu M, Gipp J, Yoon JW, Iannaccone P, Walterhouse D, Bushman W. Sonic hedgehog-responsive genes in the fetal prostate. J Biol Chem. 2009; 284: 5620-5629.
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002; 129: 4753-4761.
- Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007; 104: 5895-5900.
- Ecke I, Rosenberger A, Obenauer S, Dullin C, Aberger F, Kimmina S, et al. Cyclopamine treatment of full-blown Hh/Ptch-associated RMS partially inhibits Hh/Ptch signaling, but not tumor growth. Mol Carcinog. 2008, 47: 361-372.
- Sarangi A, Valadez JG, Rush S, Abel TW, Thompson RC, Cooper MK, et al. Targeted inhibition of the Hedgehog pathway in established malignant glioma xenografts enhances survival. Oncogene. 2009; 28: 3468-3476.
- Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol. 2010; 28: 5321-5326.
- Campbell V, Copland M. Hedgehog signaling in cancer stem cells: a focus on hematological cancers. Stem Cells Cloning. 2015; 8: 27–38.83. Campbell V, Copland M. Hedgehog signaling in cancer stem cells: a focus on hematological cancers. Stem Cells Cloning. 2015; 8: 27–38.