
Case Report
Ann Hematol Oncol. 2016; 3(9): 1113.
Triple-Double: A Triple Hit Lymphoma Hits the Kidneys Twice
Carney BJ¹, Malvar G², Freed JA¹, Pihan G², Bryke CR² and Joyce RM¹*
¹Division of Hematology & Oncology, Beth Israel Deaconess Medical Center, USA
²Department of Pathology, Beth Israel Deaconess Medical Center, USA
*Corresponding author: Joyce RM, Division of Hematology & Oncology, Beth Israel Deaconess Medical Center, Kirstein 121, 330 Brookline Avenue, Boston MA 02215, USA
Received: August 17, 2016; Accepted: October 04, 2016; Published: October 06, 2016
Abstract
Triple hit lymphoma (THL) is a rare form of non-Hodgkin lymphoma characterized by gene rearrangements involving c-MYC, BCL-2, and BCL-6. These genetic aberrations result in a promotion of growth and suppression of apoptosis, giving rise to a profoundly aggressive lymphoma. The typically rapid progression of THL often results in a high tumor burden at the time of diagnosis, and this creates a significant risk of treatment-related tumor lysis syndrome. Here, we report a case of THL that initially presented with obstructive nephropathy in the setting of spontaneous tumor lysis syndrome. To our knowledge, this is the first case reported in the literature of THL presenting in this manner.
Keywords: Triple hit lymphoma; Tumor lysis syndrome; Obstructive nephropathy
Abbreviations
BEAM: Carmustine, Etoposide, Cytarabine, and Melphalan; CNS: Central Nervous System; CT: Computed Tomography; DLBCL: Diffuse Large B- Cell Lymphoma; FDG-PET: Fludeoxyglucose- Positron Emission Tomography; R-EPOCH; THL: Triple Hit Lymphoma
Case Presentation
A previously healthy 67-year-old man presented to an outside hospital with a two week history of bilateral flank pain and acute-onset oliguria. Labs were remarkable for a serum creatinine of 1.78 mg/dL from a baseline of 0.8 mg/dL several months prior to presentation. Urinalysis was positive for leukocyte esterase although a urine culture returned with no growth. A computed tomography (CT) scan of his abdomen and pelvis revealed a 7 mm non-obstructing right renal calculus but was otherwise unremarkable. A Foley catheter was placed with good drainage of urine and the patient was started on empiric ceftriaxone and admitted for observation. His acute kidney injury was attributed to a combination of a urinary tract infection and non-steroidal anti-inflammatories that he had been using for the flank pain. Despite the above management, the patient’s renal function continued to deteriorate. Over the next six days, he became anuric and his creatinine trended up to 6.73 mg/dL on the day of transfer. During his admission at the outside hospital, labs were also notable for a steadily up-trending leukocytosis, reaching 31,500/μL with 10% band forms, and a lactate dehydrogenase (LDH) of 450 IU/L (normal range 94-250 IU/L). The patient was transferred to our center for further evaluation and management of anuric renal failure.
On admission to our hospital, labs were remarkable for urea nitrogen 68 mg/dL and creatinine 8.6 mg/dL. With the exception of mild hyponatremia and an anion gap metabolic acidosis, chemistries were otherwise unremarkable. Complete blood count revealed persistent leukocytosis to 34,700/μL. The differential was with 20% band forms and 9% atypical cells. Renal ultrasound showed moderate to severe bilateral hydronephrosis and a possible 7 mm calculus in the right ureter. Nephrology and Urology were consulted. Both consulting services felt that the patient’s presentation was most consistent with an obstructive etiology. Interventional Radiology was consulted and the patient underwent bilateral percutaneous nephrostomy tube placement. This resulted in rapid normalization of his renal function. Despite this, his leukocytosis, initially attributed to a brisk leukemoid reaction in the setting of a urinary tract infection, persisted and worsened to 51,800/μL by his second hospital day. LDH and uric acid obtained at that time returned markedly elevated at 2,422 IU/L and 17.6 mg/dL, respectively. After conferring with Nephrology, the patient was given rasburicase due to high clinical suspicion for urate nephropathy. Medical Oncology was consulted given concern for a hematologic malignancy.
Peripheral blood smear showed lymphoid cells with punchedout vacuoles and irregular nuclear contours (Figure 1). The patient underwent a bone marrow biopsy. Flow cytometry revealed a clonal population of cells expressing CD10, CD19, CD20, and CD45 comprising 52% of total events. A high grade lymphoma FISH panel was performed on uncultured mononuclear interphase peripheral blood cells (Figure 2). The IGH (14q32) and BCL-2 (18q21) dual fusion probe set showed two fusion signals positive for IGH/BCL- 2 gene rearrangement and an extra IGH signal. The MYC (8q24.1) break apart probe set showed a split signal positive for rearrangement of MYC and an extra 3' MYC signal. The BCL-6 (3q27) break apart probe set showed a split signal consistent with rearrangement of BCL-6. These rearrangements in c-MYC, BCL-2, and BCL-6 were consistent with a “triple hit” lymphoma. Metaphase chromosome analysis of circulating lymphoma cells from a direct preparation and a 24 hour unstimulated culture revealed a 46,XY,inv(3)(q25q27),t(8;14) (q24.1;q32),del(10)(p11.2), t(14;18)(q32;q21) karyotype in all 20 cells examined (Figure 3). Metaphase FISH analysis revealed that in addition to the identified chromosome rearrangements, the abnormal karyotype harbored two copies of the derivative chromosome 14 of the t(8;14)(q24.1;q32), likely resulting in further upregulation of MYC. Fludeoxyglucose-positron emission tomography (FDG-PET) scan immediately prior to initiation of treatment revealed diffuse heterogeneous FDG avidity within the bone marrow and spleen as well as extensive uptake throughout the peritoneum and surrounding the bladder.
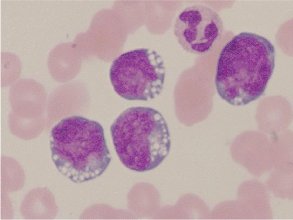
Figure 1: Wright-Giemsa stain of peripheral blood. Note lymphoid cells with
punched-out vacuoles and irregular nuclear contours.
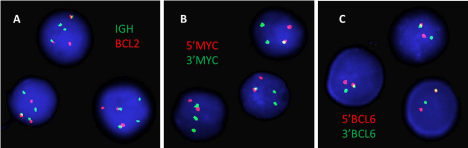
Figure 2: High grade lymphoma FISH panel performed with Abbott Molecular
probes on uncultured mononuclear interphase peripheral blood cells. A) IGH
on 14q32 (green) and BCL2 on 18q21 (red) dual fusion probe set shows two
red-green fusion signals positive for the IGH/BCL2 gene rearrangement, a
normal red BCL2 signal and two green IGH signals. B) MYC on 8q24.1 break
apart probe set shows an intact red-green MYC signal, split red 5' MYC and
green 3' MYC signals positive for rearrangement of MYC, and an extra green
3' MYC signal. C) BCL6 on 3q27 break apart probe set shows a normal
intact red-green BCL6 signal and split red 5' BCL6 and green 3' BCL6 signals
positive for rearrangement of BCL6.
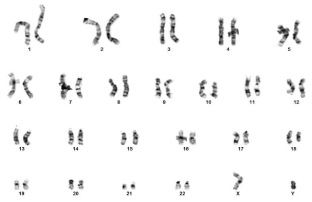
Figure 3: Metaphase chromosome analysis of circulating lymphoma cells
revealed a 46, XY,inv(3)(q25q27),t(8;14)(q24.1;q32),del(10)(p11.2),t(14;18)
(q32;q21) karyotype in all 20 cells examined.
The patient was started on dose-adjusted etoposide, prednisolone, vincristine, cyclophosphamide, doxorubicin, and subsequently received rituximab (R-EPOCH) [1]. He received three 21-day cycles of R-EPOCH. During the third cycle, he underwent lumbar puncture in anticipation of prophylactic chemotherapy. Cerebrospinal fluid (CSF) analysis returned positive for malignant cells, for which the patient received two doses intrathecal methotrexate 15 mg followed by a single dose of liposomal cytarabine 50 mg. Because of central nervous system (CNS) involvement, patient was transitioned from R-EPOCH to etoposide, ifosfamide, high-dose cytarabine, intrathecal methotrexate, and rituximab (Magrath part B, R-IVAC) [2]. On restaging imaging after three cycles R-IVAC, the patient was found to have complete resolution of his bulky disease. Bone marrow biopsy revealed normal trilineage hematopoiesis without evidence of lymphoma and cytogenetics revealed a normal male karyotype. Because of excellent clinical, radiographic, and pathologic response, the patient underwent an autologous stem cell transplant with carmustine, etoposide, cytarabine, and melphalan (BEAM) conditioning [3]. Peripheral stem cell mobilization occurred one month after completion of chemotherapy and one week after restaging bone marrow biopsy. Autologous transplant followed one week after this. Unfortunately, two months after transplant, the patient presented with aphasia and simple partial seizures which were found to be due to a CNS recurrence. Seizures continued despite treatment with high-dose systemic methotrexate, and after discussions with his family he was transitioned to comfort-focused care. The patient expired eight months after diagnosis.
Discussion and Conclusion
Triple hit lymphoma is a rare entity. In the Mitelman lymphoma database, only about 1% of lymphomas have gene rearrangements consistent with THL [4]. Like other B cell lymphomas, THL is characterized by gene rearrangements, specifically in c-MYC (t[8;14]), BCL-2 (t[14;18]), and BCL-6 (multiple potential rearrangements involving gene locus at 3q27) [5]. These “three hits” cause over-expression of c-MYC, resulting in increased cellular proliferation, protein synthesis, and metabolism, [6] and BCL-2 and BCL-6, resulting in suppression of apoptosis. Not surprisingly, the high-level expression of these three oncogenes gives rise to an extremely aggressive malignancy. Histologically, THL is similar to double hit lymphoma, having both the “starry sky” appearance of Burkitt lymphoma and the large cells with irregular nuclei and prominent nucleoli characteristic of diffuse large B cell lymphoma (DLBCL) [7]. THL is marked by an aggressive clinical course. Because it is uncommon, data regarding clinical outcomes is limited. In the largest series to date, a clinicopathologic report on 11 patients, death within 1 year of diagnosis was frequent and remission was rare despite treatment with high-dose combination chemotherapy [7]. With regards to treatment of THL, there is a similar paucity of data, although it is common for patients to received regimens that are often employed in high-grade DLBCL [8,9]. Investigations into novel therapies for multi-hit lymphomas are ongoing, as recently reviewed by Marullo and colleagues [10].
Tumor lysis syndrome is characterized by a number of electrolyte abnormalities often encountered in patients undergoing treatment for malignancy. As outlined by Cairo and Bishop, it often presents with hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia [11]. These metabolic derangements can give rise to potentially fatal complications, including acute renal failure, seizure, arrhythmia, and sudden cardiac death. Because of this, early recognition and institution of treatment with fluid resuscitation, allopurinol, and, in severe cases, rasburicase, is essential. Tumor lysis syndrome is common in patients with hematologic malignancies, with between 4 to 42% developing it at some point during their course [12]. Spontaneous tumor lysis in the absence of prior treatment is less common, having a prevalence of 1.08% based on a retrospective analysis of patients with concurrent hematologic malignancy and acute kidney injury [13]. The spontaneous lysis of tumor cells is associated with aggressive malignancies that have high tumor burdens, [14] criteria that THL frequently meets at the time of diagnosis.
Given the aggressive pathologic and clinical behavior of THL, it is not surprising that it caused spontaneous tumor lysis and resulting obstructive nephropathy in our patient. To our knowledge, this is the first documented case of THL presenting in this manner. This case highlights the importance of maintaining a high index of suspicion for a hematologic malignancy in patients presenting with obstructive nephropathy of unclear etiology in the setting of a marked leukocytosis. This suspicion, paired with a low threshold for laboratory and, if appropriate, pathologic evaluation, should allow for more rapid institution of maneuvers to manage the electrolyte derangements of tumor lysis syndrome. Subsequent diagnosis of the underlying hematologic process will allow for expedited initiation of treatment, something that is often vital in the setting of such profoundly aggressive malignancies.
References
- Wilson WH, Grossbard ML, Pittaluga S, Cole D, Pearson D, Drbohlav N, et al. Dose-adjusted EPOCH chemotherapy for untreated large B-cell lymphomas: a pharmacodynamic approach with high efficacy. Blood. 2002; 99: 2685-2693.
- Magrath I, Adde M, Shad A, Venzon D, Seibel N, Gootenberg J, et al. Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol. 1996; 14: 925-34.
- Wang EH, Chen YA, Corringham S, Bashey A, Holman P, Ball ED, et al. High-dose CEB vs. BEAM with autologous stem cell transplant in lymphoma. Bone Marrow Transplant. 2004; 34: 581-587.
- Bacher U, Haferlach T, Alpermann T, Kern W, Schnittger S, Haferlach C. Several lymphoma-specific genetic events in parallel can be found in mature B-cell neoplasms. Genes Chromosomes Cancer. 2011; 50: 43-50.
- Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003; 102: 1443-1448.
- Mationg-kalaw E, Tan LH, Tay K, Lim ST, Tang T, Lee YY, et al. Does the proliferation fraction help identify mature B cell lymphomas with double- and triple-hit translocations? Histopathology. 2012; 61: 1214-1218.
- Wang W, Hu S, Lu X, Young KH, Medeiros LJ. Triple-hit B-cell Lymphoma with MYC, BCL2, and BCL6 Translocations/Rearrangements: Clinicopathologic Features of 11 Cases. Am J Surg Pathol. 2015; 39: 1132-1139.
- Dunleavy K. Aggressive B cell Lymphoma: Optimal Therapy for MYC-positive, Double-Hit, and Triple-Hit DLBCL. Curr Treat Options Oncol. 2015; 16: 58.
- De jonge AV, Roosma TJ, Houtenbos I, Vasmel WL, van de Hem K, de Boer JP, et al. Diffuse large B-cell lymphoma with MYC gene rearrangements: Current perspective on treatment of diffuse large B-cell lymphoma with MYC gene rearrangements; case series and review of the literature. Eur J Cancer. 2016; 55: 140-146.
- Marullo R, Rutherford SC, Leonard JP, Cerchietti L. Therapeutic implication of concomitant chromosomal aberrations in patients with aggressive B-cell lymphomas. Cell Cycle. 2016; 15: 2241-2247.
- Cairo MS, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004; 127: 3-11.
- Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008; 26: 2767-2778.
- Hsu HH, Chan YL, Huang CC. Acute spontaneous tumor lysis presenting with hyperuricemic acute renal failure: clinical features and therapeutic approach. J Nephrol. 2004; 17: 50-56.
- Kekre N, Djordjevic B, Touchie C. Spontaneous tumour lysis syndrome. CMAJ. 2012; 184: 913-916.