
Special Article - Multiple Myeloma
Ann Hematol Oncol. 2016; 3(11): 1120.
Novel T Cell Therapies for Multiple Myeloma
Suen H1,2*, Bryant C1,3, Hart D3 and Joshua D1
¹Department of Haematology, Royal Prince Alfred Hospital, University of Sydney, Australia
²School of Life Sciences, University of Technology Sydney, Australia
³Dendritic Cell Research, ANZAC Research Institute, University of Sydney, Australia
*Corresponding author: Hayley Suen, Department of Haematology, Royal Prince Alfred Hospital, University of Sydney, Missenden Road, Camperdown, Sydney, NSW 2050, Australia
Received: September 30, 2016; Accepted: October 28, 2016; Published: October 31, 2016
Abstract
Multiple myeloma is a cancer of malignant plasma cells in the bone marrow and treatment has vastly improved in the last decade with the introduction of novel agents such as immunomodulatory drugs and proteasome inhibitors. New therapies are however required to eradicate residual disease and convert remission into cure. An increased understanding of immune defects in patients with myeloma has allowed for the development of immunotherapies that aim to unleash host T cells from tumour suppression and reactivate host immunity. Such T cell therapies are currently being trialled in patients producing promising results. In this review, we will describe the dysfunctional T cell landscape in myeloma, summarise the recent advances in T cell therapies and discuss future possibilities for myeloma treatment.
Keywords: Multiple myeloma; Immunotherapy; T cell; CAR-T cells; Immune checkpoint blockade; Adoptive T cell therapy
Introduction
Despite significant therapeutic gains due to the use of immunomodulatory drugs (IMiDs) and proteasome inhibitors in MM, all patients eventually relapse from persisting residual disease and MM remains incurable [1]. New therapies are required to eradicate residual disease and promote long term survival. Immunotherapy in conjunction with standard chemotherapy may be the avenue to long-term remissions and ultimately cures [2,3]. There is both direct and indirect evidence that immunological control of the malignant myeloma cells is possible. Allogeneic hematopoietic stem cell transplantation, the prototypic cellular immune therapy, can cure myeloma; however it is associated with high morbidity and mortality [4]. Indirect evidence for host anti-tumour activity includes the observation of pre-malignancy specific effector T cells in the bone marrow of patients with monoclonal gammopathy of undetermined significance (MGUS) [5], patients with disease in plateau phase, where a significant tumour load is detected but the disease remains stable and suggests a degree of host immune control [6] and the correlation between expanded CD8+ cytotoxic T cell clones and improved survival [7-10]. Whilst the immune system has the ability to target and eradicate MM tumour cells, this ability is hampered by widespread immune dysfunction in MM, which affects the T cell compartment, in particular cytotoxic T cells, which are the predominant type of effector cell involved in immune-mediated cancer destruction [11]. Tumour-induced mechanisms incapacitate immune cells, including tumour-specific cytotoxic T cells, thus permitting tumour evasion and consequent progression [12]. In this review, we will highlight the current abnormalities in the T cell landscape in MM and the development of T cell therapies for MM. In particular, we will review the use of IMiDs, checkpoint inhibitors, adoptive T cell therapy, dendritic cell (DC) vaccination and alternative approaches to MM treatment (Figure 1).
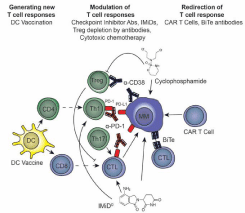
Figure 1: Overview of T cell therapies for the treatment of multiple myeloma.
Immunotherapies currently being trialled aim to enhance the host immune
response against multiple myeloma through three different methods: 1)
Generation of new T cell responses through dendritic cell (DC) vaccination to
prime T cell responses against antigens; 2) Modulation of T cell responses
by unleashing T cells from tumour suppression using checkpoint inhibitor
antibodies, administering IMiDs, removal of suppressive cells (Tregs)
and cytotoxic chemotherapy and 3) Redirection of T cell response using
genetically modified T cells such as CAR T cells and BiTe antibodies to
induce T cell recognition and killing of malignant tumour cells.
Abbreviations: Abs: Antibodies; BiTe: Bi-specific T cell engagers; CAR:
Chimeric Antigen Receptor; CTL: Cytotoxic T Lymphocyte; DC: Dendritic
Cell; IMiDs: Immunomodulatory Drugs; MM: Multiple Myeloma; PD-1:
Programmed Death Protein-1; PD-L1: PD-1 ligand; Th17: T helper 17 cells;
Treg: Regulatory T cell.
The T cell landscape in MM
A hallmark of cancer is the avoidance of immune destruction by the host immune system [13]. There are different mechanisms used by tumour cells to evade the host immune system including alterations in how T cells encounter antigen or become activated, leading to impaired cytotoxic T cell function [14,15]. Such tumourinduced immunosuppression may be partly responsible for the lack of success of immunotherapy approaches in MM in the past. A full understanding of the mechanisms of T cell suppression is required in order to restore T cell function and ultimately the ability of these T cells to mount an effective immune response against MM.
There are multiple numerical, phenotypic and functional abnormalities in the T cell compartment of MM patients [8]. One of the earliest defects detected was a decreased CD4:CD8 ratio due to a decrease in the absolute number of CD4+ T cells and a normal absolute but increased relative number of CD8 T cells [16,17]. Further decreases in this ratio have also been linked to disease progression [18].
Discrepancies in absolute numbers of regulatory T cells (Tregs) and T helper (Th) 17 cells have been reported in MM [19-24], however this may be attributed to the differences in methodology and patient selection [25]. Of more importance may be the imbalance between the numbers of Tregs and Th17 cells (Treg/Th17 ratio) as tumour progression is supported by the presence of an immune suppressive microenvironment induced by Tregs and a down regulated proinflammatory response by Th17 cells. Significantly increased Treg/ Th17 ratios have been detected in MM patients in comparison to patients with monoclonal gammopathy of undetermined significance (MGUS) and aged matched healthy controls. The increased Treg/ Th17 ratio favours a suppressive state and also correlated with a worse prognosis [25]. Conflicting reports of a skewed ratio towards Th17 cells in MM have also been described in the peripheral blood and bone marrow [26]. IL-6 and TGF-b from the surrounding BM environment plays a role in the induction of Th17 cells and a concomitant decrease in numbers of Tregs [26,27].
The transfer of membrane proteins between cells during cellto- cell contact, termed trogocytosis, can alter the phenotype and function of cells [28]. Trogocytosis occurs in MM and is more prevalent than in other B cell malignancies including chronic lymphocytic leukemia and Waldenstrom’s macroglobulinaemia [29]. These studies demonstrated T cells were more commonly recipients of trogocytosis, rather than B cells or natural killer cells. Trogocytosis occurs in a unidirectional manner and T cells acquire CD86 and human leucocyte antigen (HLA)-G antigens after contact with malignant plasma cells. The acquisition of these molecules, in particular HLA-G, alters the function of T cells, converting them into acquired Tregs, which are able to inhibit the proliferation and function of other T cells, similar to that of natural Tregs.
Numerically normal but functionally defective dendritic cells (DC) in the blood of patients can lead to impaired antigen presentation to cytotoxic T cells and suppressed recognition and killing of malignant plasma cells. After stimulation with CD40 ligand, DC from MM patients are unable to significantly upregulate the expression of the B7 co-stimulatory molecules, CD80 and CD86. The immature DCs are unable to provide co-stimulation to T cells, leading to impaired antigen presentation to T cells. The failed upregulation of DC costimulatory molecules is induced by transforming growth factor β (TGF-β) and interleukin (IL)-10 secreted by malignant plasma cells [30] and this defect can be reversed by the addition of exogenous IL- 12 or interferon (IFN)-γ [31]. Tumour secreted IL-6 can also alter the number, phenotype, function and development of DCs. IL-6 inhibits the growth of CD34+ DC progenitors, causing the cells to differentiate into CD1a- CD14+ monocytes with phagocytic function rather than antigen-presenting abilities [32]. In addition, DCs in MM also have a decreased ability to stimulate allogeneic T cell responses and these features are hallmarks of tolerogenic DC [33]. Absence of co-stimulation during T cell activation can also induce T cell anergy or hypo-responsiveness of the T cell, tolerance or T cell death. Suboptimal presentations of antigens due to the presence of inhibitory signals and/or poor co-stimulation also contribute to this anergic phenotype.
Myeloid derived suppressor cells (MDSC) are a heterogeneous population of immature myeloid progenitor cells that have tumour promoting and immune suppressive properties [34]. Increased numbers of MDSC are found in MM patients and there is a bidirectional beneficial relationship between MDSC and malignant plasma cells [35,36]. In co-cultures, MDSCs inhibit autologous T cell proliferation in both the peripheral blood and bone marrow. MDSC also exert suppressive effects on immune effector cells resulting in repressed CD4+ T cells, CD8+ T cells and NKT cell mediated antitumour immune responses [35]. The granulocytic subset of MDSC (HLA-DR- CD33+, CD11b+, CD15+) are significantly increased in MM patients with progressive disease. This subset is the most inhibitory and these cells are able to generate additional Treg cells and are stimulated by G-CSF during stem cell mobilization [36].
Expanded populations of cytotoxic CD8+ T cell clones are frequently detected in MM patients [10]. The presence of these cells is linked to a favorable prognosis [7,37,10] and are postulated to play a role in anti-tumour immunity. Despite their prognostic significance the cells are unable to proliferate in vitro in response to T cell receptor ligation, in comparison to other non-clonal T cells, suggesting tumourinduced dysfunction [38,10]. T cell clones are a universal feature of long term survivors of MM and in these patients, the clones remain proliferative suggesting a role in improving survival. [38]. Clonal T cell dysfunction was shown to be related to telomere-independent senescence or senescence associated secretory phenotype (SASP) as the cells were able to produce IFN-γ [10]. Potential targets to restore cell function, including blocking of the TGF-SMAD pathway have also been identified [10]. Restoring their function provides a unique opportunity to enhance natural anti-tumour immunity in MM and has potential to develop a novel cell therapy based on the restoration of the host’s immune response.
Standard MM therapies alter the T cell landscape
To optimize the efficacy of immunotherapy it is essential to understand the therapeutic landscape in MM, and how this alters immunity. Dramatic improvements in outcomes for MM patients over the last 15 years have been predominantly due to the advent of novel agents (namely immunomodulatory drugs and proteasome inhibitors). Three drug combinations containing two novel agents and glucocorticoids, or one novel agent, an alkylating chemotherapeutic agent and a glucocorticoid are able to induce complete responses in 75% of patients [39]. Broadly speaking, novel T cell therapies are being trialled in three clinical contexts: 1) in combination with these conventional regimens at induction or relapse, 2) after the completion of conventional therapy when disease bulk is low, or 3) in the relapsed/refractory setting when multiple lines of conventional therapy have failed. All of these strategies require an understanding of the effects of conventional therapies on T cells and how this alters their susceptibility to manipulation and in particular, suggest the window of immunological opportunity will be when disease control is at its optimum.
Immunomodulatory drugs
Immunomodulatory drugs (IMiDs) acquired this moniker for their ability to promote T cell activation. It was initially shown that Thalidomide greatly lowers the threshold for T cell proliferation and cytokine production upon TCR engagement [40]. Importantly, there is no detectable change to T cell behavior in the absence of TCR ligation, suggesting that these agents enhance T cell function, without affecting T cell specificity. Subsequent generations of IMiDs do this more potently. Mechanistically, they achieve this by bypassing the need for T cell co-stimulation, through inducing tyrosine phosphorylation of CD28 and downstream signaling [41]. The mechanism of action of IMiDs has only been recently described [42], and the T-cell stimulatory effect is attributable to cereblon-mediated modulation of the E3-ubiquitin ligase leading to the degradation of T cell suppressing signaling molecules Ikaros and Aiolos [43]. Practically, this infers that T cells can proliferate in response to the suboptimal stimulus of an immature DC, with low levels of co-stimulatory molecule expression. As DCs have a maturation defect in MM [44], demonstrating impaired up-regulation of co-stimulatory molecules, this may provide a solution in vivo to their dysfunction and impaired ability to present antigen. In T cells in CLL, there is a defect in the recruitment of signaling molecules upon TCR stimulation and subsequently an inadequate antigen-dependent F-actin polymerisation, leading to weak immunological synapses [45]. Lenalidomide has the ability to repair these defects and strengthen synapse formation in vivo [45,46] and it is possible that it supports immune synapse formation in MM.
In addition to lowering the T cell rheostat for activation, Lenalidomide has the ability to reduce the suppressive effect of Tregs and MDSCs on T effector cells, and this may further potentiate antitumour immune responses [47-49]. All of the above is demonstrated by the fact that when Lenalidomide is administered at the time of vaccination with pneumococcal 7-valent vaccine, antigen-specific T cell responses are markedly increased [50]. The effect of Lenalidomide dose and schedule has recently been examined in a murine lymphoma model [51]. Continual low dose therapy but not high dose therapy, was associated with better vaccine-related protection from tumour challenge. This was dependent on both CD4+ and CD8+ T cells, and associated with a drop in both Treg and myeloid-derived suppressor cells. Lenalidomide appears to be the most promising novel agent to take forward into clinical trials combined with DC vaccination therapies. When combined with a MM DC tumour fusion vaccine in vitro, T cell responses were enhanced, with increased Th1 polarization and killing of tumour targets [48].
IMiDs such as thalidomide are also able to stimulate additional T cell clones, which are linked to a better prognosis. In the MM6 trial that investigated the use of thalidomide as maintenance therapy following autologous transplant, T cell clones were initially detected in 48% of patients prior to transplant. After transplant, for patients who received control maintenance (prednisolone), the T cell clone incidence remained similar, at 47%, however, in the patient group that received thalidomide maintenance, clone incidence increased to 76% [9]. The presence of clones on multivariate analysis was found to be an independent factor in prolonging progression free survival.
Cytotoxic chemotherapy: The most commonly used cytotoxic chemotherapy agent in the treatment of MM in transplant eligible patients is cyclophosphamide and it has stimulated recent interest for its immunomodulatory effects. The effects of cyclophosphamide are dose dependent. The immune modulatory effects occur at a low dose (20 mg/kg) in mice which promote immune-mediated suppression of tumour growth [52]. It does this at least in part through the depletion of Tregs [53]. However, at higher doses (200 mg/kg) there is bone marrow suppression and decreases in effector cell numbers, and these benefits are lost. The immune effects of cyclophosphamide are also dose dependent in humans [54]. Importantly, the doses of cyclophosphamide used in MM in induction therapies like cyclophosphamide, thalidomide and dexamethasone (CTD) are similar to the doses found to be immune-potentiating (300 mg/m2) in humans [55,56].
Glucocorticoids: While novel agents and cyclophosphamide may have positive effects on T cell mediated immunity, Glucocorticoids (GC) are ubiquitous in MM treatment protocols, and they dramatically suppress T cell immunity. T cells undergo apoptosis at pharmacological concentrations of GC, dependent on levels of BCL-2 [57]. In addition, GC modulate gene expression through glucocorticoid response elements, altering Tec kinase levels, and suppressing signaling downstream of the T cell receptor [58].
Therefore, conventional therapeutic regimens used in MM contain agents that both enhance and inhibit T cell immunity. In particular, with regards to GC, a difficult balance must be struck between the synergistic anti-MM effects of GC and novel agents, and the fact that GC will greatly limit any immune potentiating effects. A rational way to assess this is in the maintenance phase after autologous transplantation, with a randomization to IMIDs with or without GC, and this is currently being assessed in the context of pomalidomide (MM14).
Antibody therapies
Immune checkpoint blockade in MM: Cancers escape immune surveillance by suppressing anti-tumour T cells through multiple mechanisms, including the hijacking of immune checkpoints pathways involving molecules such as programmed cell death protein-1 (PD-1) and cytotoxic T lymphocyte associated antigen-4 (CTLA-4) [59]. PD-1 is a particularly relevant checkpoint in MM as there is increased expression on CD4, CD8 and NK cells from patients with MM [60,61]. Interestingly, PD-1 levels normalize upon recovery after autologous stem cell transplantation [60]. However, PD-1 is not expressed on clonally expanded cytotoxic T cells in the MM bone marrow [62]. These T cell clones are associated with an improved prognosis [37] [63,10], hence are strong candidates as MM-specific T cells. For immune checkpoint blockade to be an effective modality, it is not only crucial to be able to reverse tumour-induced T cell dysfunction through blockade, but also for those re-activated T cells to have the ability to specifically recognize a tumour epitope. The lack of PD-1 on the prognostically significant clonal T cells may explain the limited clinical response to PD-1 blockade in MM [62].
In addition, PD-L1, a ligand for PD-1, is increased on malignant plasma cells from MM patients in comparison to healthy plasma cells [61], as well as on the supportive stromal cells [64], although there are also conflicting reports of low levels of PD-L1 on plasma cells [65].
When mice are transplanted with murine MM, PD-1 is unregulated on CD4 and CD8 T cells. This is proportional to tumour load, is more marked in tumour-bearing tissue and is associated with an exhausted phenotype [66]. Furthermore, PD-L1 expression was driven in part by increased copy number and was highest in hyperdiploid MM, and not associated with poor prognostic cytogenetics subgroups. Pre-clinical data supports the promise of PD-1 checkpoint blockade. Anti-PD-1 increases granzyme expression in T and NK cells from refractory MM patients and enhances their ability to kill autologous MM cells. The addition of PD-1 blockade enhanced T cell responses to a myeloma DC fusion vaccine in vitro, with increased Th1 polarization and cytotoxic capacity [60]. Taken together, this means the PD-1/PDL-1 axis is relevant in MM, and has the capacity to enhance autologous anti-MM immunity.
The initial results of single agent PD-1 checkpoint blockade in MM were disappointing [62,67]. Of the 27 relapsed and refractory MM patients who received 2nd weekly nivolumab, there was only one response. However, given all patients had received at least 2 prior lines of therapy, and 52% had received at least 4 prior lines, the fact that 63% achieved stable disease with a median PFS of 10 weeks, may indicate some limited activity in this heavily pre-treated group [67]. It is not possible to draw much from a single patient response in a clinical trial, but the one individual, who responded required radiotherapy to a single rib plasmacytoma mid study, and recommenced nivolumab afterwards. They remain in CR at 14 months since discontinuation. There is current interest in the abscopal effect, where by local radiotherapy leads to the regression of distant tumours through immunological mechanisms, and checkpoint blockade promotes this [68,69]. An intriguing study of immunological biomarkers in a melanoma patient who developed the abscopal effect after receiving radiotherapy whilst on CTLA4 blockade (ipilumumab), demonstrated an increase in CD4 T cell activation and melanoma antigen specific T cell responses with CTLA4 blockade, but with no further increase after radiotherapy despite a dramatic clinical response [68]. Further studies are required to dissect the mechanisms behind this phenomenon.
In contrast to the limited efficacy of single agent PD-1 blockade, more impressive effects have been described in combination with IMiDs. It has recently been demonstrated that lenalidomide can enhance immune checkpoint blockade-induced immune responses in MM [70]. Pembrolizumab in combination with lenalidomide and low dose dexamethasone was tested in relapsed patients with 51% having received >3 prior therapies, and 41% having failed IMiDs [71]. The ORR was 76% (n=17), with 4 VGPR and 9 PR. Whilst responses in IMiD refractory patients are mentioned, the response rate in lenalidomide refractory patients is not mentioned. A combination of pembrolizumab, pomalidomide and low dose dexamethasone has also been tested in the heavily pre-treated patients with a median of 3 prior lines of therapy, all patients having received both IMiDs and Proteasome inhibitors, and 75% refractory to both, with 21% refractory to Lenalidomide alone. In addition, all patients had cytogenetic abnormalities, with 1q+ in 72% and high-risk FISH in 40%. The ORR was subsequently lower at 50% (11/22) with 23% VGPR or better and 27% PR [72]. However, these studies are uncontrolled, and ORR to low dose dexamethasone and pomalidomide in the relapsed refractory setting are significant; for example the ORR was 35% in IFM2009-02 with a more heavily pretreated population with a median of 5 prior lines of therapy [73], and 34% in a similarly heavily pre-treated population with a median of 3 prior lines of therapy [74]. Larger cohorts and controlled trials are required, to confirm what appears to be synergism between these two immunomodulatory therapies.
BiTe antibodies: Another application of antibody therapy to treat MM is through Bi-specific T cell engagers (BiTe). BiTe antibodies are composed of single fragment chain variable components with two specificities, joined by a linker [75]. This allows one antibody to target one antigen on one cell, and a different antigen on another cell. The widest application of this is to target an antigen that the tumour bears, and the T cell receptor, through CD3 [75]. Importantly, this not only brings the T cell and tumour within proximity, but leads to tumour-specific activation of T cells [76], and the formation of an immunological synapse [77].
Clinical efficacy has been demonstrated in ALL with a CD3-CD19 directed BiTe, Blinotumumab, in relapsed disease [78], with 43% CR rates. There is pre-clinical validation of this approach in MM. B cell maturation antigen (BCMA), is an attractive target as its expression is restricted to MM cells, and it is expressed widely in MM [79]. Subsequently, one CD3-BCMA targeting BiTe has been shown to induce MM cell line death and promote survival in Xenograft models [80], and an alternative construct kills MM cell lines, while activating T cells and releasing inflammatory cytokines, with favorable pharmacokinetics in non-human primate studies with no reported major toxicity [81]. The great benefit of antibody therapeutics is how prone they are to alterations and optimization. A BiTe antibody targeting CD138 and CD3 with a high affinity IgG1 region to target Fc and therefore associate with activate NK cells in addition to T cells, has shown promise in preclinical testing [82]. These antibodies now need to be tested in the clinic and this is currently under commercial investigation. The most promising agent targets BCMA, CD3 and has enhanced Fc binding, and is currently under investigation in a Phase I clinical trial (NCT02064387).
Immune effects of anti-CD38 antibodies: MM targeting antibodies have had dramatic effects in the clinic, with the use of daratumumab, a CD38 specific antibody that has single agent efficacy [83] and also induces deep responses in combination with other novel agents [84]. However, CD38 is expressed on numerous immune cell populations and anti-CD38 antibodies are immunomodulatory. Daratumumab has been shown to selectively abrogate a novel population of CD38+ Tregs, which are more immunosuppressive than CD38- Tregs in vitro. Concomitantly, there was an increase in absolute numbers of helper and cytotoxic T cell subsets. Interestingly, daratumumab promoted clonal T cell expansions, and this was more prevalent in patients with good clinical responses, suggesting an antigen driven T cell response with daratumumab treatment [85].
Cellular therapies
DC vaccination: Dendritic cells are the professional antigenpresenting cells of the immune system, and have the ability to stimulate T cell responses to new tumour antigens. When used as a cellular therapy, this involves (1) removing cells (usually monocytes) from the body, (2) maturing them in vitro into monocyte-derived DC (MoDC) (3) exposing them to a source of tumour antigen and (4) re administering them so that they will traffic to lymph nodes and prime T cell responses [86]. There was a scientific rationale for this approach in MM, as myeloid DC (mDC) have an altered phenotype in MM with partial maturation [32,87,88] and a maturation defect, with impaired up-regulation of CD80 when stimulated by CD40L [30]. This is attributable to factors within the tumour microenvironment including TGFβ and IL-10, and can be reversed in vitro with IL-12 and IFN-γ [31]. Consequently, mDC have a decreased ability to stimulate T cell proliferation and the production of Th1 cytokines [32,87,88]. Hence, it was reasonable to perform DC maturation outside of this suppressive tumour microenvironment. While MoDC generated in the presence of MM tumour lysate or MM bone marrow sera are defective [32,89,90], MoDC developed from MM patients’ monocytes in the absence of tumour-related material are functional [91,92]. However, there are reservations as to how well Mo-DC migrate after injection and also in their ability to process and present antigen [93,94].
Numerous trials have now been conducted using DC vaccination in MM and a full discussion is beyond the scope of this review but definitive clinical efficacy has been difficult to demonstrate. The Mayo group reported a survival advantage with their DC enriched mononuclear cell vaccine of 5.3 years when compared to a historical control cohort with 3.4 years, however this is not truly controlled data and remains open to criticism [95]. Rosenblatt et al vaccinated patients with a MoDC tumour fusion as a consolidation strategy after conventional induction and autologous transplantation, and while they report 24% converting from PR to CR during the vaccination schedule [96], paraprotein levels continue to decrease in up to 39% of patients more than 100 days after autologous transplant without any further intervention [97], so it is unclear whether this was truly a vaccine effect. However, we must remember the lessons learnt from the clinical evaluation of Sipeleucel T, the only FDA-accredited blood DC based vaccine therapy in use [98]. It was necessary to enroll 512 patients with castrate-resistant prostate cancer in a randomized controlled trial to demonstrate a benefit, and furthermore, while there was no different in progression-free survival, there was a significant increase in overall survival of 2 to 3 months. Hence, the nature of immunological control is fundamentally different to chemotherapy induced tumour death, with the potential to enter a state of equilibrium, wherein tumour persists but its effects are limited. This demands that we assess the clinical effects of immune therapies in a fundamentally different way to conventional agents and small molecule inhibitors, with an increased focus on survival benefits and disease stability, along with the use of robust harmonised T cell biomarkers. Indeed, when we view the MM DC vaccination trials in this light, multiple trials have reported stable disease in vaccinated patients with demonstrable anti-tumour T cell responses [99,100,101], and this may reflect clinical efficacy.
The successful induction of anti-tumour T cell responses in vivo in these trials demonstrates that MM patients’ T cells can recognize the tumour if appropriately primed. There are options for improving DC vaccination, notably by using blood DC as an alternative source [102]. There are a number of therapies which can enhance T cell responses, for example PD-1 checkpoint blockade enhances the potency of a MM DC vaccine in vitro [60], and is now being assessed in a clinical trial (NCT01067287). Moving forward, it will be sensible to rationally combine DC therapies with T cell therapies for improved clinical outcomes.
Chimeric antigen receptor (CAR) T cells in MM: The fundamental role of the cytotoxic T cell in eliminating tumour cells has recently been underscored by advances in immunotherapy using chimeric antigen receptor (CAR) T cells and immune checkpoint blockade [103]. CAR T cells are autologous T cells that are genetically modified to express receptors containing an antigen recognition domain that recognises a specific target on tumour cells, as well as co-stimulatory and T cell activation domains. Infusion of CAR T cells aims to induce T cell recognition and killing of malignant tumour cells. A number of CAR T cells against different targets are currently being investigated in MM patients. CD19 CAR T cells have been successful in inducing good clinical responses in a number of B cell malignancies, most noticeably in acute lymphoblastic leukemia. Interestingly, CD19 CAR T cells have also been efficacious MM despite the absence of this marker on plasma cells [104]. Autologous stem cell transplantation followed by infusion of CD19 CAR T cells led to a complete response in a MM patient who was previously refractory to nine lines of therapy. There was no detectable serum or urine monoclonal protein and the response persisted despite the fact that CAR T cells were no longer detectable. The efficacy was attributed to CAR T cells targeting a CD19+ MM precursor plasma cell population or to the possible depletion of normal CD19+ B cells that were pro-myeloma [105].
Anti-myeloma activity has also been demonstrated with CAR T cells recognizing BCMA which is expressed by some B cells, normal plasma cells and malignant plasma cells [79]. Two patients were treated with the highest dose level of BCMA-CAR T cells (9x106 CAR+ T cells/kg body weight). The first patient had chemotherapy resistant disease with a heavily infiltrated BM of up to 90% plasma cells. Following administration of BCMA-CAR T cells, plasma cells were undetectable by flow cytometry and the patient went into stringent complete remission for 17 weeks prior to relapse. The second patient, who also had chemotherapy resistant disease with plasma cells constituting 80% of the BM, has had a very good partial response to date following CAR T cell treatment. Higher numbers of BCMA-CAR T cells were required to induce responses in patients in comparison to CD19-CAR T cells, which could possibly be attributed to weaker BCMA expression on plasma cells, differences in CAR T cell design and the presence of soluble BCMA in serum and in the bone marrow. The BCMA-CAR T cells were found to be of an effector memory T cell phenotype and expressed increased levels of PD-1 and CD57, indicative of a highly differentiated cell phenotype [106]. Both patients treated on the highest dose level exhibited cytokine release syndrome and experienced fevers, hypotension, dyspnoea and cytopenias, much like the patients that received CD19-CAR T cell treatment in MM and ALL. Other targets for are also currently being investigated, including CD138, a molecule that is highly expressed on MM plasma cells, with stable responses observed in 5 patients thus far [107].
Despite the impressive clinical results thus far, the mechanisms of action and efficacy are not completely understood. In addition, adoptively-transferred T cells are also autologous T cells, which may also mean they are susceptible to existing tumour suppressive mechanisms [108]. Preclinical data demonstrated that the presence of Tregs in the tumour microenvironment could inhibit the CAR T cell activity against tumour cells in a murine model [109]. Modifications to the early generation of CAR T cells to secrete IL-12 were required to combat the suppressive effects exerted by Tregs [110]. Further problems may include off target issues, as target antigens may be expressed on normal non-malignant cells or tissues. B cell aplasia was seen in patients with the use of CD19 CAR T cells [111]. Unexpected serious adverse events and fatalities toxicity against cardiac tissue resulted from MAGE-A3 CAR T cells as cardiac muscle protein, titin, was found to be a previously unknown alternative target for the MAGE-3 CAR T cells [112].
Future possibilities
Although MM remains incurable at present, there are a wide range of treatment options that reduce tumour burden and improve the quality of life for patients. The overall survival for MM has improved dramatically since the introduction of novel therapies such as IMiDs and proteasome inhibitors, however most patients eventually relapse. The T cell therapies discussed in this review now provide a promising avenue towards a potential cure for patients with myeloma by targeting and removing residual tumour burden, however, there is still much to learn about their long term persistence. T cell therapies, which utilise autologous T cells, may also be susceptible to the same in vivo tumour suppressive mechanisms and tumour escape can occur through clonal evolution and loss of antigenicity [108]. The re-activation of innate, natural tumour immunity in MM patients remains a prospect to eradicate residual tumour cells and produce a cure. The restoration of protective tumour-induced T cell clones may be a possible avenue to achieve this and also for the identification of MM-specific antigens that could be exploited for therapy.
References
- Paiva B, van Dongen JJ, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015; 125: 3059-3068.
- Reichardt VL, Okada CY, Liso A, Benike CJ, Stockerl -Goldstein KE, Engleman EG, et al. Idiotype vaccination using dendritic cells after autologous peripheral blood stem cell transplantation for multiple myeloma - a feasibility study. Blood. 1999; 93: 2411-2419.
- Danylesko I, Beider K, Shimoni A, Nagler, A. Novel Strategies for Immunotherapy in Multiple Myeloma: Previous Experience and Future Directions. Clinical and Developmental Immunology. 2012; 28.
- Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, et al. Treatment of multiple myeloma. Blood. 2004; 103: 20-32.
- Dhodapkar MV, Krasovsky J, Osman K, Geller MD. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J Exp Med. 2003; 198: 1753-1757.
- Joshua DE, Brown RD, Gibson J. Multiple myeloma: why does the disease escape from plateau phase? Br J Haematol. 1994; 88: 667-671.
- Brown RD, Yuen E, Nelson M, Gibson J, Joshua D. The prognostic significance of T cell receptor beta gene rearrangements and idiotype-reactive T cells in multiple myeloma. Leukemia. 1997; 11: 1312-1317.
- Raitakari M, Brown RD, Gibson J, Joshua DE. T cells in myeloma. Hematol Oncol. 2003; 21: 33-42.
- Brown R, Spencer A, Ho PJ, Kennedy N, Kabani K, Yang S, et al. Prognostically significant cytotoxic T cell clones are stimulated after thalidomide therapy in patients with multiple myeloma. Leuk Lymphoma. 2009; 50: 1860-1864.
- Suen H, Brown R, Yang S, Weatherburn C, Ho PJ, Woodland N, et al. Multiple myeloma causes clonal T cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016; 30: 1716-1724.
- Maher J, Davies ET. Targeting cytotoxic T lymphocytes for cancer immunotherapy. Br J Cancer. 2004; 91: 817-821.
- Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007; 121: 1-14.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-674.
- Crespo J, Sun H, Welling TH, Tian Z, Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current opinion in Immunology. 2013; 25: 214-221.
- Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014; 35: 51-60.
- Mellstedt H, Holm G, Pettersson D, Björkholm M, Johansson B, Lindemalm C, et al. T cells in monoclonal gammopathies. Scand J Haematol. 1982; 29: 57-64.
- Mills KH, Cawley JC. Abnormal monoclonal antibody-defined helper/suppressor T-cell subpopulations in multiple myeloma: relationship to treatment and clinical stage. Br J Haematol. 1983 53: 271-275.
- Kay NE, Leong TL, Bone N, Vesole DH, Greipp PR, Van Ness B, et al. Blood levels of immune cells predict survival in myeloma patients: results of an Eastern Cooperative Oncology Group phase 3 trial for newly diagnosed multiple myeloma patients. Blood. 2001; 98: 23-28.
- Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood. 2006; 107: 3940-3949.
- Beyer M, Schultze JL. Regulatory T cells in cancer. Blood. 2006; 108: 804-811.
- Prabhala RH, Neri P, Bae JE, Tassone P, Shammas MA, Allam CK, et al. Dysfunctional T regulatory cells in multiple myeloma. Blood. 2006; 107: 301-304.
- Feyler S, von Lilienfeld-Toal M, Jarmin S, Marles L, Rawstron A, Ashcrof AJ, et al. CD4(+)CD25(+)FoxP3(+) regulatory T cells are increased whilst CD3(+)CD4(-)CD8(-)alphabetaTCR(+) Double Negative T cells are decreased in the peripheral blood of patients with multiple myeloma which correlates with disease burden. Br J Haematol. 2009; 144: 686-695.
- Gupta R, Ganeshan P, Hakim M, Verma R, Sharma A, Kumar L. Significantly reduced regulatory T cell population in patients with untreated multiple myeloma. Leuk Res. 2011; 35: 874-878.
- Braga WM, Atanackovic D, Colleoni GW. The role of regulatory T cells and TH17 cells in multiple myeloma. Clin Dev Immunol. 2012; 2012: 293479.
- Favaloro J, Brown R, Aklilu E, Yang S, Suen H, Hart D, et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk Lymphoma. 2014; 55: 1090-1098.
- Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, et al. Elevated IL -17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood. 2010; 115: 5385-5392.
- Noonan K, Borrello I. The immune microenvironment of myeloma. Cancer Microenviron. 2011; 4: 313-323.
- Joly E, Hudrisier D. What is trogocytosis and what is its purpose? Nat Immunol. 2003; 4: 815.
- Brown R, Kabani K, Favaloro J, Yang S, Ho PJ, Gibson J, et al. CD86+ or HLA-G+ can be transferred via trogocytosis from myeloma cells to T cells and are associated with poor prognosis. Blood. 2012; 120: 2055-2063.
- Brown R, Pope B, Murray A, Esdale W, Sze DM, Gibson J, et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood. 2001. 98: 2992-2998.
- Brown R, Murray A, Pope B, Sze DM, Gibson J, Ho PJ, et al. Either interleukin-12 or interferon gamma can correct the dendritic cell defect induced by transforming growth factor beta in patients with myeloma. British Journal of Haematology. 2004; 125: 743-748.
- Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B, et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood. 2002; 100: 230-237.
- Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006; 108: 1435-1440.
- Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006; 108: 1435-1440.
- Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010; 40: 2969-2975.
- Görgün GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood. 2013; 121: 2975-2987.
- Favaloro J, Liyadipitiya T, Brown R, Yang S, Suen H, Woodland N, et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk Lymphoma. 2014; 55: 2893-2900.
- Brown RD, Spencer A, Ho PJ, Kennedy N, Kabani K, Yang S, et al. Prognostically significant cytotoxic T cell clones are stimulated after thalidomide therapy in patients with multiple myeloma. Leuk Lymphoma. 2009; 50: 1860-1864.
- Bryant C, Suen H, Brown R, Yang S, Favaloro J, Aklilu E, et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013; 3: e148.
- Mailankody S, Korde N, Lesokhin AM, Lendvai N, Hassoun H, Stetler-Stevenson M, et al. Minimal residual disease in multiple myeloma: bringing the bench to the bedside. Nat Rev Clin Oncol. 2015; 12: 286-295.
- Haslett PA, Corral LG, Albert M, Kaplan G. Thalidomide costimulates primary human T lymphocytes, preferentially inducing proliferation, cytokine production, and cytotoxic responses in the CD8+ subset. J Exp Med. 1998; 187: 1885-1892.
- LeBlanc R, Hideshima T, Catley LP, Shringarpure R, Burger R, Mitsiades N, et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood. 2004; 103: 1787-1790.
- Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012; 26: 2326-2335.
- Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agent’s lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4 (CRBN.). Br J Haematol. 2014; 164: 811-821.
- Brown RD, Pope B, Murray A, Esdale W, Sze DM, Gibson J, et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood. 2001; 98: 2992-2998.
- Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008; 118: 2427-2437.
- Shanafelt TD, Ramsay AG, Zent CS, Leis JF, Tun HW, Call TG, et al. Long-term repair of T-cell synapse activity in a phase II trial of chemoimmunotherapy followed by lenalidomide consolidation in previously untreated chronic lymphocytic leukemia (CLL). Blood. 2013; 121: 4137-4141.
- Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. 2009; 58: 1033-1045.
- Luptakova K, Rosenblatt J, Glotzbecker B, Mills H, Stroopinsky D, Kufe T, et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol Immunother. 2013; 62: 39-49.
- De Keersmaecker B, Fostier K, Corthals J, Wilgenhof S, Heirman C, Aerts JL, et al. Immunomodulatory drugs improve the immune environment for dendritic cell-based immunotherapy in multiple myeloma patients after autologous stem cell transplantation. Cancer Immunol Immunother. 2014; 63: 1023-1036.
- Noonan K, Rudraraju L, Ferguson A, Emerling A, Pasetti MF, Huff CA, et al. Lenalidomide-induced immunomodulation in multiple myeloma: impact on vaccines and antitumor responses. Clin Cancer Res. 2012; 18: 1426-1434.
- Sakamaki I, Kwak LW, Cha SC, Yi Q, Lerman B, Chen J, et al. Lenalidomide enhances the protective effect of a therapeutic vaccine and reverses immune suppression in mice bearing established lymphomas. Leukemia. 2014; 28: 329-337.
- Motoyoshi Y, Kaminoda K, Saitoh O, Hamasaki K, Nakao K, Ishii N, et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep. 2006; 16: 141-146.
- Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004; 34: 336-344.
- Bass KK, Mastrangelo MJ. Immunopotentiation with low-dose cyclophosphamide in the active specific immunotherapy of cancer. Cancer Immunol Immunother. 1998; 47: 1-12.
- Berd D, Mastrangelo MJ, Engstrom PF, Paul A, Maguire H. Augmentation of the human immune response by cyclophosphamide. Cancer Res. 1982; 42: 4862-4866.
- Hoon DS, Foshag LJ, Nizze AS, Bohman R, Morton DL. Suppressor cell activity in a randomized trial of patients receiving active specific immunotherapy with melanoma cell vaccine and low dosages of cyclophosphamide. Cancer Res. 1990; 50: 5358-5364.
- Gilardini Montani MS, Tuosto L, Giliberti R, Stefanini L, Cundari E, Piccolella E. Dexamethasone induces apoptosis in human T cell clones expressing low levels of Bcl-2. Cell Death Diffe.1999; 6: 79-86.
- Petrillo MG, Fettucciari K, Montuschi P, Ronchetti S, Cari L, Migliorati G, et al. Transcriptional regulation of kinases downstream of the T cell receptor: another immunomodulatory mechanism of glucocorticoids. BMC Pharmacol Toxicol. 2014; 15: 35.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12: 252-264.
- Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother. 2011; 34: 409-418.
- Gorgun G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res. 2015; 21: 4607-4618.
- Suen H, Brown R, Yang S, Ho PJ, Gibson J, Joshua D. The failure of immune checkpoint blockade in multiple myeloma with PD-1 inhibitors in a phase 1 study. Leukemia. 2015; 29: 1621-1622.
- Bryant C, Suen H, Brown R, Yang S, Favaloro J, Aklilu E, et al. Long-term survival in multiple myeloma is associated with a distinct immunological profile, which includes proliferative cytotoxic T-cell clones and a favourable Treg/Th17 balance. Blood Cancer J. 2013; 3: e148.
- Ray A, Das DS, Song Y, Richardson P, Munshi NC, Chauhan D, et al. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia. 2015; 29: 1441-1444.
- Paiva B, Azpilikueta A, Puig N, Ocio EM, Sharma R. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015; 29: 2110-2113.
- Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant. 2011; 17: 1133-1145.
- Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol. 2016; 34: 2698-2704.
- Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012; 366: 925-931.
- Park SS, Dong H, Liu X, Harrington SM, Krco CJ, Grams MP, et al. PD-1 Restrains Radiotherapy-Induced Abscopal Effect. Cancer Immunol Res. 2015; 3: 610-619.
- Görgün G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin Cancer Res. 2015; 21: 4607-4618.
- San Miguel J, Mateos MV, Shah JJ, Ocio EM, Rodriguez-Otero P, Reece D, et al. A Phase II Study of Anti PD-1 Antibody Pembrolizumab, Pomalidomide and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood. 2015; 126: 506.
- Leleu X, Attal M, Arnulf B, Moreau P, Traulle C, Michalet M, et al. High Response Rates to Pomalidomide and Dexamethasone in Patients with Refractory Myeloma, Final Analysis of IFM 2009-02. Blood. 2011; 118: 812.
- Lacy MQ, Kumar SK, LaPlant BR, Laumann K, Gertz MA, Hayman SR, et al. Pomalidomide Plus Low-Dose Dexamethasone (Pom/Dex) in Relapsed Myeloma: Long Term Follow up and Factors Predicing Outcome in 345 Patients. Blood. 2012; 120: 201.
- Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015; 93: 290-296.
- Brischwein K, Parr L, Pflanz S, Volkland J, Lumsden J, Klinger M, et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J Immunother. 2007; 30: 798-807.
- Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006; 43: 763-771.
- Topp MS, Gökbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015; 16: 57-66.
- Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013; 19: 2048-2060.
- Hipp S, Deegen P, Wahl J, Blanset D, Thomas O, Rattel B, et al. BI 836909, a Novel Bispecific T Cell Engager for the Treatment of Multiple Myeloma Induces Highly Specific and Efficacious Lysis of Multiple Myeloma Cells In Vitro and Shows Anti-Tumor Activity In Vivo. Blood. 2015; 126: 2999.
- Vu MD, Moser S, Delon C, Latzko M, Gianotti R, Lüoend R, et al. A New Class of T-Cell Bispecific Antibodies for the Treatment of Multiple Myeloma, Binding to B Cell Maturation Antigen and CD3 and Showing Potent, Specific Antitumor Activity in Myeloma Cells and Long Duration of Action in Cynomolgus Monkeys. Blood. 2015; 126: 2998.
- Zou J, Chen D, Zong Y, Ye S, Tang J. Immunotherapy based on bispecific T-cell engager with hIgG1 Fc sequence as a new therapeutic strategy in multiple myeloma. Cancer Sci. 2015; 106: 512-521.
- Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N Engl J Med. 2015; 373: 1207-1219.
- Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N Engl J Med. 2016; 375: 754-766.
- Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016; 128: 384-394.
- Anguille S, Smits EL, Bryant C, Van Acker HH, Goossens H, Lion E, et al. Dendritic Cells as Pharmacological Tools for Cancer Immunotherapy. Pharmacol Rev. 2015; 67: 731-753.
- Brimnes MK, Svane IM, Johnsen HE. Impaired functionality and phenotypic profile of dendritic cells from patients with multiple myeloma. Clin Exp Immunol. 2006; 144: 76-84.
- Martin-Ayuso M, Almeida J, Perez-Andres M, Cuello R, Galende J, Gonzalez-Fraile MI, et al. Peripheral blood dendritic cell subsets from patients with monoclonal gammopathies show an abnormal distribution and are functionally impaired. Oncologist. 2008; 13: 82-92.
- Hayashi T, Hideshima T, Akiyama M, Raje N, Richardson P, Chauhan D, et al. Ex vivo induction of multiple myeloma-specific cytotoxic T lymphocytes. Blood. 2003; 102: 1435-1442.
- Yang DH, Park JS, Jin CJ, Kang HK, Nam JH, Rhee JH, et al. The dysfunction and abnormal signaling pathway of dendritic cells loaded by tumor antigen can be overcome by neutralizing VEGF in multiple myeloma. Leuk Res. 2009; 33: 665-670.
- Pfeiffer S, Gooding RP, Apperley JF, Goldschmidt H, Samson D. Dendritic cells generated from the blood of patients with multiple myeloma are phenotypically and functionally identical to those similarly produced from healthy donors. Br J Haematol. 1997; 98: 973-982.
- Raje N, Gong J, Chauhan D, Teoh G, Avigan D, Wu Z, et al. Bone marrow and peripheral blood dendritic cells from patients with multiple myeloma are phenotypically and functionally normal despite the detection of Kaposi's sarcoma herpesvirus gene sequences. Blood. 1999; 93: 1487-1495.
- De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003; 63: 12-17.
- Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM., et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009; 15: 2531-2540.
- Lacy MQ, Mandrekar S, Dispenzieri A, Hayman S, Kumar S, Buadi F, et al. Idiotype-pulsed antigen-presenting cells following autologous transplantation for multiple myeloma may be associated with prolonged survival. Am J Hematol. 2009; 84: 799-802.
- Rosenblatt J, Avivi I, Vasir B, Uhl L, Munshi NC, Katz T, et al. Vaccination with dendritic cell/tumor fusions following autologous stem cell transplant induces immunologic and clinical responses in multiple myeloma patients. Clin Cancer Res, 2013; 19: 3640-3648.
- Gonsalves WI, Gertz MA, Dispenzieri A, Lacy MQ, Lin Y, Singh PP, et al. Implications of continued response after autologous stem cell transplantation for multiple myeloma. Blood. 2013; 122: 1746-1749.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010; 363: 411-422.
- Curti A, Tosi P, Comoli P, Terragna C, Ferri E, Cellini C, et al. Phase I/II clinical trial of sequential subcutaneous and intravenous delivery of dendritic cell vaccination for refractory multiple myeloma using patient-specific tumour idiotype protein or idiotype (VDJ)-derived class I-restricted peptides. Br J Haematol. 2007; 139: 415-424.
- Yi Q, Szmania S, Freeman J, Qian J, Rosen NA, Viswamitra S, et al. Optimizing dendritic cell-based immunotherapy in multiple myeloma: intranodal injections of idiotype-pulsed CD40 ligand-matured vaccines led to induction of type-1 and cytotoxic T-cell immune responses in patients. Br J Haematol. 2010; 150: 554-564.
- Rosenblatt J, Vasir B, Uhl L, Blotta S, Macnamara C, Somaiya P, et al. Vaccination with dendritic cell/tumor fusion cells results in cellular and humoral antitumor immune responses in patients with multiple myeloma. Blood. 2011; 117: 393-402.
- Fromm PD, Papadimitrious MS, Hsu JL, Van Kooten Losio N, Verma ND, Lo TH, et al. CMRF-56(+) blood dendritic cells loaded with mRNA induce effective antigen-specific cytotoxic T-lymphocyte responses. Oncoimmunology. 2016; 5: e1168555.
- Ikeda H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int Immunol. 2016; 28: 349-353.
- Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015; 373: 1040-1047.
- Garfall AL, Stadtmauer EA, June CH. Chimeric Antigen Receptor T Cells in Myeloma. N Engl J Med. 2016; 374: 193-194.
- Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti -B-cell- maturation-antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood, epub ahead of print. 2016.
- Guo B, Chen M, Han Q, Hui F, Dai H, Zhang W, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma. Journal of Cellular Immunotherapy. 2016; 2: 28-35.
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015; 21: 687-692.
- Lee JC, Hayman E, Pegram HJ, Santos E, Heller G, Sadelain M, et al. In vivo inhibition of human CD19-targeted effector T cells by natural T regulatory cells in a xenotransplant murine model of B cell malignancy. Cancer Res. 2011; 71: 2871-2881.
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012; 119: 4133-4141.
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371: 1507-1517.
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-Derived HLA-A1–Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3–Directed T Cells. Sci Transl Med. 2013; 5: 197ra103.
- Keats JJ, Chesi M, Egan JB, Garbitt VM, Palme, SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012; 120: 1067-1076.