
Research Article
Ann Hematol Oncol. 2017; 4(1): 1132.
Functional Assessment of Myeloid-Derived Suppressor Cells, Mesenchymal Stromal Cells, and Regulatory T Cells for the Control of T Cell Function: Implications for Graft-versus- Host Disease
Siegmund DM¹, Schäfer I¹, Koch R¹, Singh A¹, Handgretinger R¹, Rieber N1,2, Hartl D1,3 and Mezger M¹*
¹Department of General Paediatrics, Haematology and Oncology, University Children’s Hospital Tübingen, Germany
²Department of Pediatrics, Kinderklinik Muenchen Schwabing, Klinikum Schwabing, StKM GmbH und Klinikum rechts der Isar, Technical University of Munich, Germany
³Roche Pharma Research & Early Development (pRED), Immunology, Inflammation and Infectious Diseases (I3) Discovery and Translational Area, Roche Innovation Center Basel, Switzerland
*Corresponding author: Mezger M, Department of General Paediatrics, Haematology and Oncology, University Children’s Hospital Tübingen, Hoppe-Seyler- Str. 1, 72076 Tübingen, Germany
Received: December 22, 2016; Accepted: February 11, 2017; Published: February 14, 2017
Abstract
Uncontrolled T cell responses cause harm in various diseases, which lead to cell-based therapeutic approaches to dampen T cell activation, including mesenchymal stromal cells (MSCs), regulatory T cells (Tregs) and myeloidderived suppressor cells (MDSCs). One major application is graft-versus-host disease (GvHD), a severe complication caused by alloreactive T cells in patients undergoing allogenic stem cell transplantation (alloSCT). Human MSCs are already used for the treatment of GvHD, however, MSCs have to be expanded and their clinical benefit still remains unclear. Therefore, we systematically compared the functional capacity of Tregs, polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) and MSCs to suppress alloreactive T cell responses. Freshly isolated PMN-MDSCs showed the strongest inhibition of T cell proliferation compared to MSCs and Tregs, but the available cell number was limited. Thus, we generated cytokine-induced PMN-MDSCs from peripheral blood mononuclear cells (PBMCs) and from bone marrow mononuclear cells (BMMCs) in vitro. BMMC-derived PMN-MDSCs effectively suppressed T cell proliferation and dampened secretion of Interferon-γ, while PMN-MDSCs generated from PBMCs showed weaker inhibition. The effects of BMMC-derived PMN-MDSCs were partially dependent on cell contact similar to freshly isolated PMN-MDSCs. In conclusion, generated PMN-MDSCs from bone marrow might represent a novel cellular therapeutic to dampen excessive T cell responses for the management of GvHD.
Abbreviations
alloSCT: allogenic Stem Cell Transplantation; APC: Allophycocyanin; BM: Bone Marrow; BMMCs: Bone Marrow Mononuclear Cells; CFSE: Carboxyfluorescein Succinimidyl (diacetate) Ester; FITC: Fluorescein Isothiocyanate; GM-CSF: Granulocyte-Macrophage Colony-Stimulating Factor; HLA-DR: Human Leucocyte Antigen D-related; Ig: Immunoglobulin; IFNγ: Interferon-γ; IL: Interleukin; MSCs: Mesenchymal Stromal Cells; MDSCs: Myeloid-Derived Suppressor Cells; PerCP: Peridinin Chlorophyll; PBMCs: Peripheral Blood Mononuclear Cells; PE: Phycoerythrin; PMN-MDSCs: Polymorphonuclear MDSCs; Stim: Stimulated; Tregs: Regulatory T cells; TGF-ß: Transforming Growth Factor-ß
Introduction
Unbalanced T cell responses drive a variety of disease pathologies, ranging from autoimmune diseases to graft-versus-host disease (GvHD) [1,2]. Beyond immunosuppressants, several cell types with T cell suppressive effects have been investigated for cellbased therapeutic applications, such as mesenchymal stromal cells (MSCs), myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs). But so far, the T cell suppressive capacity of these cell types has not been systematically compared side-by-side. For many hematological diseases, such as leukemia, allogenic stem cell transplantation (alloSCT) is a potentially curative approach, however, limited by the life-threatening complication of GvHD [1]. Transplant conditioning increases danger signals and inflammatory cytokines, followed by activation of alloreactive T cells, leading to tissue damage and further boosting the disease [1,3]. In first clinical trials for the treatment of GvHD patients, cell-based therapies with MSCs [4,5] as well as freshly isolated or in vitro expanded Tregs [6,7] showed feasibility, safety and encouraging outcomes. Furthermore, either BM-derived or in vitro generated MDSCs were found to inhibit GvHD in mice [8,9].
MSCs are multipotent cells that can differentiate into mesenchymal cell lineages. MSCs are found in several human tissues, such as bone marrow (BM), umbilical cord blood or adipose tissue [10]. During in vitro expansion, MSCs are plastic adherent and have a fibroblastic appearance. Human MSCs express CD73, CD90, and CD105 as surface molecules, but not CD34, CD45 and human leucocyte antigen D- related (HLA-DR). MSCs can modulate immune function of various cells, for example T cells, B cells, and natural killer cells [4,11,12].
MDSCs represent a heterogeneous population of immature cells from myeloid cell lineage [13]. MDCSs are functionally defined by their T cell suppressive capacity and are further sub-divided into two subsets. In humans, polymorphonuclear (PMN-) MDSCs express CD11b+, CD66b+, CD14-, and monocytic MDSCs are CD14+, and CD15-[14]. Several groups presented different methods to generate MDSCs in vitro from murine BM cells as well as human peripheral blood mononuclear cells (PBMCs) and BM cells [8,9,15,16].
Tregs are a subset of T lymphocytes and play a central role for the immunological self-tolerance as well as for the control of undesired immune reactions [17]. Tregs are characterized as CD4+CD25highFoxP3+[17]. In contrast to activated effector T cells, no exclusive Treg-specific cell marker is available so far and hence, the isolation of Tregs is still problematic [17,18].
In order to investigate which human cell type, MSCs, PMNMDSCs or Tregs, displays the greatest potential for T cell suppressive therapies, we systematically compared their immunomodulatory effects towards T cells side-by-side. We analyzed the capacity of each cell type to suppress T cell proliferation and the release of Interferon-γ (IFNγ). Furthermore, we analyzed if cell contact is required for the inhibitory effect and if the number of available cells is sufficient for a potential clinical application.
Materials and Methods
Isolation and expansion of human MSCs
Human MSCs were derived from excessive material of standard bone marrow biopsies. Excess material was used after informed consent in accordance with the Declaration of Helsinki and approval by the University Children’s Hospital Tübingen’s IRB (Institutional Review Board [IRB] approval 338/2013 B02). MSCs were cultured in the GMP facility at the Department of General Paediatrics, Haematology/Oncology in Tübingen using animal serum-free medium as described previously [5,19]. In brief, 10-15 ml bone marrow (BM) aspirates of healthy donors were resuspended in DMEM medium (1 g/l glucose, Lonza, Basel, Switzerland) supplemented with 80 IU/ml heparin sulfate, 1 mM L-glutamine (both from Biochrom, Berlin, Germany) and 108/ml irradiated human platelets (University of Tübingen, blood donor center). After 2-3 days of incubation at 37°C and 10 % CO2, non-adherent cells were removed. MSCs were expanded over a period of 3-4 weeks and harvested using TrypLE Select (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). Microbial analyses was performed regularly and the purity (>95%) of MSCs was defined by flow cytometry on the basis of CD73, CD105, CD45, and CD3 as well as CD14 (to exclude T cells and monocytes, respectively) (all antibodies from BD Biosciences, San Jose, CA, USA). A characterization of MSCs is shown in the supplement (Supplementary Figures S1,S2).
Isolation of PMN-MDSCs and CD4+CD25+ Tregs
PBMCs were prepared from heparinized peripheral blood of healthy volunteers or buffy coats (Blood bank Tuebingen, Germany) by density gradient centrifugation with Biocoll separating solution (Biochrom) and washed twice with RPMI-1640 medium (Biochrom). Cell viability was checked by dye exclusion of Trypan blue staining solution (Sigma-Aldrich, St. Louis, MO, USA).
PMN-MDSCs were isolated based on previously established methods [20,21]. Briefly, PMN-MDSCs were obtained from the PBMC fraction by labelling with anti-CD66b fluorescein isothiocyanate (FITC) antibodies and two sequential positive selections with anti- FITC MicroBeads (all Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s protocol. CD4+CD25+ Tregs were isolated from the PBMC fraction by using CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec), according to the manufacturer’s protocol. The purity of all isolated cells was >95% as confirmed by flow cytometry. A characterization of PMN-MDSCs and Tregs is shown in the supplement (Supplementary Figures S3,S4).
In vitro generation of cytokine-induced MDSCs
PBMCs or bone marrow mononuclear cells (BMMCs) were isolated by density gradient centrifugation and cultured at 37°C, 5% CO2 with RPMI-1640 (Biochrom) supplemented with 10% FCS (Gibco, Thermo Fi Scientific), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Biochrom). Cell density was 5x105 PBMC or 3x105 BMMCs/ml. Cells were stimulated with 10 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Genzyme, Cambridge, MA, USA) or in combination of 10 ng/ml GMCSF with 10 ng/ml Interleukin-6 (IL-6; Miltenyi Biotec). Medium and supplements were refreshed every 3-4 days. After incubation for 7 days, adherent cells were removed by Detachin (Genlantis, San Diego, CA, USA). For functional assays, cytokine-induced MDSCs were isolated with CD33 MicroBeads, human (Miltenyi Biotec), according to the manufacturer’s protocol. The purity of isolated cells was >90%, as assessed by flow cytometry. A characterization of the cytokineinduced MDSCs is shown in the supplement (Supplementary Figures S5,S6).
Characterization by flow cytometry
First, cells were isolated and if required, cultured as described above. Cells were washed with phosphate- buffered saline (Sigma- Aldrich), incubated with antibodies for 15 min at room temperature, again washed and analyzed by flow cytometry with FACSCaliburTM (BD Biosciences). MSCs were stained with anti- human CD34-FITC, CD90-Phycoerythrin (PE), CD105-FITC, CD45-FITC (Miltenyi Biotec), CD73-PE, CD271-PE and HLA-ABC-PE (BD Biosciences). PMN-MDSCs were stained with anti-human CD66b-PE, CD11b- Allophycocyanin (APC),CD33-PE, CD14-APC, CXCR4-APC, HLA-DR-Peridinin chlorophyll (PerCP) (Miltenyi Biotec) and CD16-PerCP (BioLegend, San Diego, CA, USA). CD4+CD25+Tregs were stained with anti-human CD45RA-FITC, CD3-PerCP, CD4- APC, CD25-PE, CD127-FITC and FoxP3-APC (Miltenyi Biotec). Cytokine-stimulated CD33+ MDSCs from PBMCs and from BMMCs were stained with anti-human CD14-FITC, CD66b-FITC, CD56- FITC (BD Biosciences), CD33-PE, HLA-DR-PerCP, CD11b-APC, CD3-PerCP, CXCR4-APC (Miltenyi Biotec), CD16-PerCP, CD19- PE, CCR5-PE, and CCR2-APC (BioLegend). Mouse immunoglobulin (Ig) M-FITC, Mouse IgG2a-FITC, Mouse IgG2b-FITC, Mouse IgG1- PE, REA Control (S)-PE, Rat IgG2b-APC, Mouse IgG1-APC, Mouse IgG2a-APC, Mouse IgG2a-PerCP (Miltenyi Biotec), Mouse IgG1- FITC, Mouse IgG2a-PE, Mouse IgG1-PerCP (BD Biosciences) were used as isotype controls. All experiments were at least performed three times in independent experiments.
T cell suppression assay
Responder PBMCs were obtained from healthy volunteers and stained with Vybrant CFDA SE Cell Tracer Kit (CFSE: carboxyfluorescein succinimidyl (diacetate) ester) according to manufacturer’s protocol (Life Technologies). PBMCs were stimulated with 100 IU/ml Interleukin-2 and 1 mg/ml muromonab- CD3 (OKT3) (Janssen-Cilag, Neuss, Germany). In standardized way, 60,000 responder PBMCs per well in a 96-well microtiter plate were co-cultured with 10.000 (ratio 1:0.16), 15.000 (ratio 1:0.25), or 30.000 (ratio 1:0.5) immunomodulatory cells at 37°C and 5% CO2. As a positive control, stimulated responder PBMCs without immunomodulatory cells were used, and as a negative control, unstimulated PBMCs were analyzed. In case of co-culture with MSCs, seeding of MSCs was performed the day before responder PBMCs were added. Except of supplementary figure S7 and S8, all experiments were performed in an allogenic setting. The cell culture media was RPMI-1640 containing 10% donor- specific human serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Biochrom).
After co-culture for 4-5 days, supernatants were collected and frozen at -80°C for cytokine analysis. Cells were harvested and stained with anti-CD4 PE and anti-CD8a APC (BioLegend). Only propidium iodide (BD Biosciences)-negative cells were considered for analysis. To determine polyclonal T cell proliferation, the fluorescence intensity of CFSE from gated CD4+ and CD8+ T cells was analyzed by flow cytometry. Polyclonal T cell proliferation was normalized to responder PBMC without any immunomodulatory cells as 100%. Where indicated, responder PBMCs and immunosuppressive cells were separated by a semipermeable membrane with 0.4 μm pores in a transwell plate (Corning, New York, USA) in order to investigate if cell-to-cell contact is required. In transwell experiments, responder PBMCs and immunomodulatory cells were seeded in two ratios (1:0.16 and 1:0.5) without cell-to-cell contact. All experiments were at least performed three times in independent experiments.
Cytokine analysis
Supernatants from T cell suppression assays were taken on day 4 or 5 and analyzed by using IFN-γ DuoSet ELISA (R&D systems, Abingdon, United Kingdom), according to manufacturer’s protocol.
Statistical analysis
Data are reported as means ± standard derivations (SD).
Statistical analysis was performed by using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA). Differences between the groups were determined by a Mann-Whitney test regarding non- Gaussian distribution. In all tests, a P value =0.05 was considered to be significant (*P = 0.05, **P =0.01, ***P =0.001).
Results
PMN-MDSCs show stronger suppression of T cell proliferation than MSCs and CD4+CD25+ Tregs To investigate the suppressive potential of different immunomodulatory cells, we systematically performed CFSE assays to analyze polyclonal T cell proliferation and checked release of IFNγ by enzyme-linked immunosorbent assay (ELISA). Beforehand, we characterized all immunomodulatory cells by flow cytometry (Supplementary Figures S2-S6). MSCs, PMNMDSCs and CD4+CD25+ Tregs strongly decreased proliferation of T cells in a dose-dependent manner (Figure 1A). PMN-MDSCs suppressed both CD4+ and CD8+ T cell proliferation significantly stronger than MSCs (P=0.009 and P=0.002, respectively) and freshly isolated CD4+CD25+ Tregs (P=0.008 and P=0.04, respectively). At a ratio of 1:0.5, 96.3% of CD4+ and 94.3% of CD8+ T cell proliferation was inhibited by PMN-MDSCs, 85.8% of CD4+ and 83.9% of CD8+ T cell proliferation by MSCs and 91.5% of CD4+ and 88.8% of CD8+ T cell proliferation by Tregs, respectively. In an autologous experimental setting, the inhibitory effect of PMN-MDSCs was decreased compared to the allogenic setting, but in the same range as MSCs and Tregs (Supplementary Figure S7). Surprisingly, MSCs showed a significantly greater inhibition of IFNγ release than PMNMDSCs (P=0.01) and CD4+CD25+ Tregs (P=0.05) implying that different mechanisms for immunomodulation are utilized (Figure 1B). Furthermore, we analyzed the inhibitory effect of MSCs and PMN-MDSCs from the same donors with similar results compared to different donors of the cell types (Supplementary Figure S8).
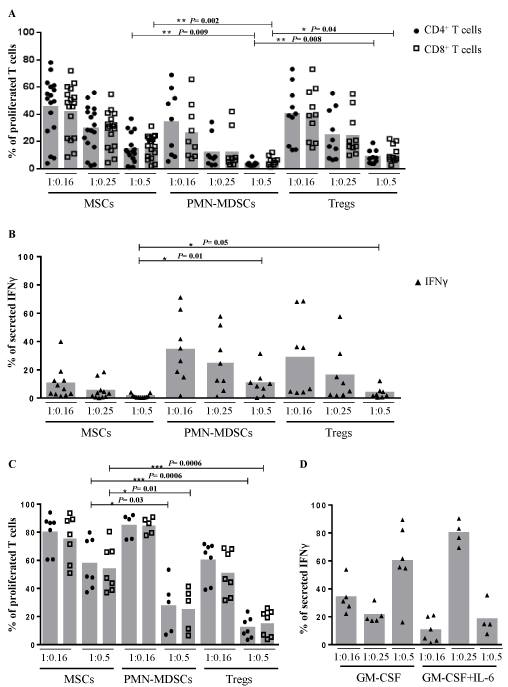
Figure 1: Suppressive effects on T cell proliferation and secretion of Interferon-γ by MSCs, polymorphonuclear MDSCs and Tregs. (A) Responder PBMCs were
labeled with CFSE, stimulated with OKT3 and IL-2 to polyclonal T cell proliferation and cultured in different ratios of 1:0.16, 1:0.25 or 1:0.5 with the indicated
immunomodulatory cells. After 4 days of incubation, the proliferation of T cells was analyzed by flow cytometry. Each donor of responder PBMCs is presented by ●
and for CD4+ and by ? for CD8+ T cells. MSCs of 9 different donors, PMN-MDSCs of 7 donors and CD4+CD25+ Tregs of 6 donors were examined. (B) After coculture
the supernatants were collected and determined by IFNγ ELISA. Each ? indicates the level of IFNγ from a single donor of responder PBMCs after co-culture
with the indicated cell type from the same assays as in 1A. (C) PBMCs and immunomodulatory cells were used in ratios of 1:0.16 and 1:0.5 and the cells were
separated by a semipermeable membrane in a transwell system during T cell suppression assay. Each donor of responder PBMCs is presented by ● and for CD4+
and by ? for CD8+ T cells. MSCs of 4 different donors, PMN-MDSCs of 5 donors and CD4+CD25+ Tregs of 4 donors were examined. (D) The supernatants of the
stimulated PBMCs were collected after 4 days incubation in a transwell system and analyzed by IFNγ ELISA. All measurements were normalized to the control of
untreated PBMCs as 100%. Each indicates the level of IFNγ from a single donor of responder PBMCs after co-culture with the indicated cell type.
Cell contact is required for effective suppression of T cells
In order to analyze if cell-to-cell contact is required for the inhibitory effect, stimulated responder PBMCs and immunomodulatory cells were separated by a semipermeable membrane in a transwell system (Figure 1C). MSCs only slightly suppressed T cell proliferation showing that cell-to-cell contact is required for effective immunosuppression. At the same time, the level of secreted IFNγ clearly decreased in the transwell system (Figure 1D). PMN-MDSCs inhibited both T cell proliferation and release of IFNγ in this experimental setting, where the higher cell number had a much stronger effect than the lower. Furthermore, we detected in the transwell system a suppression of T cell proliferation and IFNγ release by CD4+CD25+ Tregs.
In vitro generated PMN-MDSCs derived from PBMCs
For treatment of patients, the amount of immunoregulatory cells is crucial. Both CD4+CD25+ Tregs and PMN-MDSCs are rare in the peripheral blood of healthy humans. We isolated these cells by magnetic separation from PBMCs with a percentage of 0.46±0.23% and 0.37±0.16%, respectively (Table 1). These cell numbers are too low for clinical applications. Therefore, we tried to generate MDSCs out of PBMCs by cytokine stimulation to increase the cell numbers. Based on the protocols from Lechner, et al. [15], we stimulated PBMCs with 10 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) alone or in combination with 10 ng/ml IL-6 for 7 days and afterwards selected CD33+ MDSCs. Both types of stimulated CD33+ PMN-MDSCs derived from PBMCs suppressed proliferation of allogenic CD4+ and CD8+ T cells in a dose-dependent manner, but to a lesser extent than detected by Lechner, et al. [15] in the autologous T cell setting (Figure 2A). This shows that in vitro generated CD33+ MDSCs have lower T cell suppressive effects than the isolated PMNMDSCs from fresh blood. Furthermore, we observed a concentrationdependent decrease of secreted IFNγ in the supernatants (Figure 2B) and an inhibition of T cell proliferation and secretion of IFNγ in the transwell system (Figure 2C,D). No difference was found between GM-CSF and GM-CSF with IL-6 stimulated CD33+ MDSCs. Taken together, the reduced immunosuppressive effect as well as the low cell number of CD33+ MDSCs from PBMCs (Table 1) limits any clinical application at the moment.
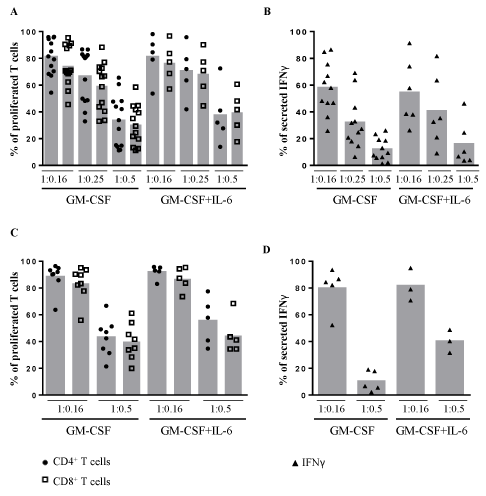
Figure 2: Suppressive effects on T cell proliferation and secretion of Interferon-γ by PBMC-derived cytokine-induced CD33+ MDSCs. (A) PBMCs were isolated and
cultured with GM-CSF alone or in Combination with IL-6 for 7 days followed by CD33+ magnetic selection of cytokine-induced CD33+ MDSCs. On day 7, responder
PBMCs from another healthy donor were isolated, labeled with CFSE and stimulated with OKT3 and IL-2. These PBMCs were co-cultured with cytokine-induced
CD33+ MDSCs from PBMCs in different ratios of 1:0.16, 1:0.25 or 1:0.5. After 4 days of incubation, the T cell proliferation was assessed by flow cytometry. Each
donor of responder PBMCs is presented by ● and for CD4+ and by ? for CD8+ T cells. CD33+ MDSCs of 10 different PBMC donors stimulated with GM-CSF alone
and CD33+ MDSCs of 4 PBMC donors stimulated with GM-CSF and IL-6 were examined. (B) After co-culture, the supernatants were collected and analyzed by
IFNγ ELISA. Each ? indicates the level of IFNγ from a single donor of responder PBMCs after co-culture with the indicated cell type. (C,D) Same procedures as
in A and B beside cells were cultured in a transwell system without cell-to-cell contact in ratio 1:0.16 or 1:0.5. All measurements were normalized to the control of
untreated PBMCs as 100 %. Each donor of responder PBMCs is presented by ● and for CD4+ and by ? for CD8+ T cells or by for the level of IFNγ. CD33+ MDSCs
of 6 different PBMC donors stimulated with GM-CSF alone and CD33+MDSCs of 3 PBMC donors stimulated with GM-CSF and IL-6 were examined.
![]()
Isolated cell numbers (in percent of PBMCs or BMMCs)
Mean ± SD
PMN-MDSCs
0.37 ± 0.16
CD4+CD25+ Tregs
0.46 ± 0.23
GM-CSF stimulated PMN-MDSCs derived from PBMCs
0.40 ± 0.16
GM-CSF and IL-6 stimulated PMN-MDSCs derived from PBMCs
0.37 ± 0.28
GM-CSF stimulated PMN-MDSC derived from BMMCs
1.01 ± 0.48
GM-CSF and IL-6 stimulated PMN-MDSC derived from BMMCs
0.96 ± 0.33
Table 1: Percentage of isolated cells.
In vitro generated PMN-MDSCs derived from BMMCs
Next, we isolated BMMCs and stimulated again with either GMCSF alone or GM-CSF and IL-6 to generate more immature CD33+ MDSCs. These cells showed significantly greater inhibition of T cell proliferation (P=0.0006 at CD4+ T cells and P=0.0008 at CD8+ T cells) and IFNγ release (P=0.006) compared to CD33+ MDSCs from cytokine-stimulated PBMCs (Figure 3A,B). The inhibitory effect of BMMCs stimulated with GM-CSF and IL-6 in combination was slightly smaller compared to cells with GM-CSF alone. In the transwell system, CD33+MDSCs from BMMCs also suppressed T cell proliferation and secretion of IFNγ in a dose-dependent manner (Figure 3C,D).
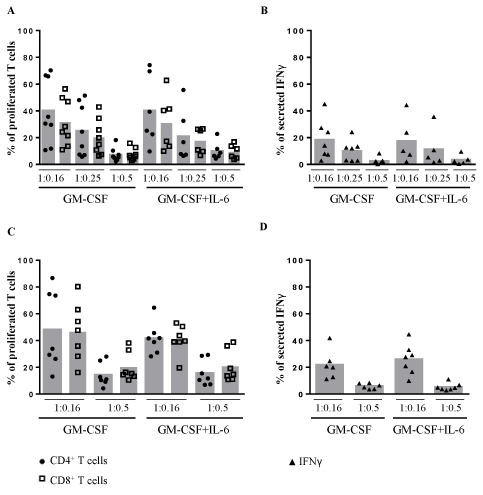
Figure 3: Suppressive effects on T cell proliferation and secretion of Interferon-γ by BMMC-derived cytokine-induced CD33+ MDSCs. (A) BMMCs were isolated
and cultured for 7 days with either GM- CSF or GM-CSF and IL-6 followed by CD33+ selection of cytokine-induced MDSCs. On day 7, responder PBMCs from
another healthy donor were isolated, labeled with CFSE and stimulated with OKT3 and IL-2. These PBMCs and CD33+ MDSCs from BMMCs were co-cultured in
different ratios of 1:0.16, 1:0.25 or 1:0.5. After 4 days of incubation the T cell proliferation was determined by flow cytometry. Each donor of responder PBMCs is
presented by ● and for CD4+ and by ? for CD8+ T cells. CD33+MDSCs of 6 different BMMC donors stimulated with GM-CSF alone and CD33+ MDSCs of 5 BMMC
donors stimulated with GM-CSF and IL-6 were examined. (B) After co-culture, the supernatants were collected and analyzed by IFNγ ELISA. Each ? indicates
the level of IFNγ from a single donor of responder PBMCs after co-culture with the indicated cell type. (C,D) Same procedures as in A and B, however, cells were
cultured in a transwell system without cell-to-cell contact in ratio 1:0.16 or 1:0.5. All measurements were normalized to the control of untreated PBMCs as 100%.
Each donor of responder PBMCs is presented by ● and for CD4+ and by ? for CD8+ T cells or by for the level of IFNγ. CD33+MDSCs of 5 different BMMC donors
stimulated with GM-CSF alone and CD33+ MDSCs of 5 BMMC donors stimulated with GM-CSF and IL-6 were examined.
Besides the suppressive effect, the amount of immunoregulatory cells represents a key issue for potential later clinical application. After cell culture and magnetic isolation, the available cell number of CD33+ MDSCs out of PBMCs is around 0.4%, which is in the same range as directly isolated PMN-MDSCs (Table 1). The amount of CD33+ MDSCs out of BMMCs reaches 1.0% (Table 1), which indicates a higher amount of MDSCs and their precursors in the bone marrow than in the peripheral blood.
Discussion
Uncontrolled T cell responses cause harm in a variety of diseases, such as GvHD, providing the rationale to develop cell-based therapeutic approaches to control overshooting T cell activation, including Tregs, MSCs and MDSCs. Previous studies showed promising results of different cell types in vitro and in vivo for the treatment of GvHD [5, 7-9].
Here, we systematically and comprehensively compared the immunomodulatory effects of human MSCs, PMN-MDSCs and CD4+CD25+ Tregs. Various mechanisms how MSCs can interact with immune cells have been reported, such as transforming growth factor-ß (TGF-ß), hepatocytce growth factor, prostaglandin E2, indoleamine-2,3-dioxygenase, interleukin-10, galectin-1, programmed death-ligand 1 pathway, Notch pathway as well as expression of adhesion molecules [22-24]. The immunosuppressive function of MDSCs includes for example depletion of L-arginine, cysteine and tryptophane, production of nitric oxide, reactive oxygen species as well as peroxynitrite and release of anti-inflammatory cytokines [13,25]. Tregs also use several mechanisms to suppress immune cells, such as galectin-1, TGF-ß, IL-10, IL-35, cytolytic molecules as Granzyme B and F as and via cytotoxic T lymphocyte antigen 4 [17,26,27]. We detected the highest suppression of allogenic T cell proliferation by freshly isolated PMN-MDSCs and by bone marrow-derived CD33+ cells after GM-CSF stimulation. Interestingly, however, MSCs suppressed the secretion of IFNγ during co-culture more potent than PMN-MDSCs and Tregs did. Even without cell-cell contact, all three cell types inhibited T cell proliferation and release of IFNγ. The inhibition mediated by CD4+CD25+ Tregs in the transwell system was unexpectedly strong since it has been reported that Tregs require direct cell contact [28, 29]. Based on previous publications, we speculate that infectious tolerance took place in the lower compartment of the transwell containing allogenic stimulated PBMCs mediated by cytokines from Tregs in the upper transwell [30,31]. One alternative explanation could be that during magnetic separation of CD4+CD25+ Tregs, it isn’t possible to differentiate between cells with high or intermediate expression of CD25; therefore isolated cells consist of a heterogeneous population of CD4+ T cells, including type 1 Tregs and Th3. These cells display immunosuppressive effects towards T cells by soluble factors, such as IL-10 and TGF-ß [32]. The particularly high immunosuppressive capacity of PMN-MDSCs increased our interest, but we detected only 0.5% of these cells in peripheral blood of healthy humans, which is similar to other groups [13]. Subsequently, the cytokine-induced CD33+ MDSCs generated from PBMCs showed a lower suppression of T cell proliferation and IFNγ-secretion compared to the results of Lechner, et al. [15]. In addition, we noticed a suppressive effect generated by CD33+ MDSCs by direct cell contact as well as in the transwell system without cell contact. The main reason for these discrepancies could be due to the fact that we used allogenic PBMCs and stimulated them for T cell proliferation, whereas Lechner, et al. [15] used autologous T cells, an issue requiring further investigations. Moreover, the moderate inhibition of T cell proliferation could be due to the already matured state of the CD33+ MDSCs derived from PBMCs. Taken together, the immunosuppressive capacity of PBMCderived CD33+ MDSCs was moderate in our allogenic setting and the available cell numbers were too low for clinical applicability. Further cytokines or chemokines need to be tested for their potential to generate MDSCs in vitro, for example, cytokines from the IL-1 family [20]. Combinations of several factors could also increase the induction of MDSCs, such as GM-CSF with IL-6 and Finasteride [33].
The generated CD33+ MDSCs from human BMMCs suppressed T cell proliferation and secretion of IFNγ more efficiently than CD33+ MDSCs generated from PBMCs, both with cell-to-cell contact as well as without direct contact. The inhibition of T cell proliferation by BMMC-derived CD33+ MDSCs was more prominent in our settings than previously demonstrated by Marigo, et al. [16]. However, we had a lower cellular yield after incubation and CD33+ MDSCs isolation with around only 1% of the initial BMMCs compared to their 70- 80% recovery without any selection. We used the same concentration of cytokines (10 ng/ml GM-CSF± IL-6) for the induction of MDSCs from PBMCs or BMMCs and incubated cells for 7 days according to Lechner, et al. [15]. However, these concentrations were distinctly lower as the concentration used by Marigo, et al. [16] (each 40 ng/ml) as well as the incubation time was longer and we selected the CD33+ MDSCs from stimulated BMMCs by magnetic separation, providing a potential explanation for these discrepant findings. Overall, BMMCderived CD33+ MDSCs showed robust immunosuppressive capacity encouraging to search for further factors to increase their cellular yield. Furthermore, a combination of several immunosuppressive cell types, such as MSCs and CD33+ MDSCs could represent a promising therapeutic approach, similar as shown previously for MSCs combined with Tregs in mice [34].
In summary, we systematically compared the immunosuppressive capacity of several human immune cell types. Freshly isolated PMNMDSCs as well as in vitro generated CD33+ MDSCs derived from bone marrow showed the strongest inhibition of T cell proliferation. After further optimizations, particularly regarding cellular yield, these studies may pave the way towards new cellular therapies for the treatment of T cell mediated diseases, such as GvHD.
Acknowledgment
This work was supported by grants from the José Carreras Leukämie-Stiftung, Germany [14/01] and the Stefan-Morsch- Stiftung, Germany.
References
- Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012; 12: 443-458.
- Galgani M, De Rosa V, Matarese G. T cell metabolism and susceptibility to autoimmune diseases. Mol Immunol. 2015; 68: 558-563.
- Markey KA, MacDonald KPA, Hill GR. The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood. 2014;124: 354-362.
- Amorin B, Alegretti AP, Valim V, Pezzi A, Laureano AM, da Silva MAL, et al. Mesenchymal stem cell therapy and acute graft-versus-host disease: a review. Hum Cell. 2014; 27:137-150.
- Müller I, Kordowich S, Holzwarth C, Isensee G, Lang P, Neunhoeffer F, et al. Application of multipotent mesenchymal stromal cells in pediatric patients following allogeneic stem cell transplantation. Blood Cells, Mol Dis. 2008; 40: 25-32.
- Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011; 117: 3921-3928.
- Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. 2011; 117: 1061-1070.
- Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1–dependent mechanism that is up-regulated by interleukin-13. Blood. 2010; 116: 5738-5747.
- Messmann JJ, Reisser T, Leithäuser F, Lutz MB, Debatin KM, Strauss G. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood. 2015; 126: 1138-1148.
- Bernardo ME, Pagliara D, Locatelli F. Mesenchymal stromal cell therapy: a revolution in Regenerative Medicine[quest]. Bone Marrow Transplant. 2012; 47:164-171.
- Maitra B, Szekely E, Gjini K, Laughlin MJ, Dennis J, Haynesworth SE, et al. Human mesenchymal stem cells support unrelated donor hematopoietic stem cells and suppress T cell activation. Bone Marrow Transplant. 2004; 33: 597-604.
- Tabera S, Pérez-Simón JA, Díez-Campelo M, Sánchez-Abarca LI, Blanco B, López A, et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica. 2008; 93: 1301-1309.
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009; 9:162-174.
- Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150.
- Lechner MG, Liebertz DJ, Epstein AL. Characterization of Cytokine-Induced Myeloid-Derived Suppressor Cells from Normal Human Peripheral Blood Mononuclear Cells. The Journal of Immunology. 2010;185: 2273-2284.
- Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-Induced Tolerance and Immune Suppression Depend on the C/EBPβ Transcription Factor. Immunity. 2010; 32: 790-802.
- Safinia N, Leech J, Hernandez-Fuentes M, Lechler R, Lombardi G. Promoting transplantation tolerance; adoptive regulatory T cell therapy. Clin Exp Immunol. 2013;172: 158-168.
- Gregori S, Passerini L, Roncarolo MG. Clinical outlook for type-1 and FOXP3+ T regulatory cell-based therapy. Front Immunol. 2015; 6: 593.
- Müller I, Kordowich S, Holzwarth C, Spano C, Isensee G, Staiber A, et al. Animal serum-free culture conditions for isolation and expansion of multipotent mesenchymal stromal cells from human BM. Cytotherapy. 2006; 8: 437-444.
- Ballbach M, Hall T, Brand A, Neri D, Singh A, Schaefer I, et al. Induction of Myeloid-Derived Suppressor Cells in Cryopyrin-Associated Periodic Syndromes. J Innate Immun. 2016; 8: 493-506.
- Rieber N, Wecker I, Neri D, Fuchs K, Schafer I, Brand A, et al. Extracorporeal photopheresis increases neutrophilic myeloid-derived suppressor cells in patients with GvHD. Bone Marrow Transplant. 2014; 49: 545-552.
- Gieseke F, Böhringer J, Bussolari R, Dominici M, Handgretinger R, Müller I. Human multipotent mesenchymal stromal cells use galectin-1 to inhibit immune effector cells. Blood. 2010; 116: 3770-3779.
- Bernardo ME, Fibbe WE. Mesenchymal stromal cells and hematopoietic stem cell transplantation. Immunol Lett. 2015; 168: 215-221.
- Castro-Manrreza ME, Montesinos JJ. Immunoregulation by Mesenchymal Stem Cells: Biological Aspects and Clinical Applications. J Immunol Res. 2015; 2015: 394917.
- Botta C, Gullà A, Correale P, Tagliaferri P, Tassone P. Myeloid derived suppressor cells in multiple myeloma: preclinical research and translational opportunities. Front Oncol. 2014; 4:348.
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010; 10: 490-500.
- Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T cell suppression – a diverse arsenal for a moving target. Immunology. 2008; 124: 13-22.
- Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998; 10: 1969-1980.
- Thornton AM, Shevach EM. CD4(+)CD25(+) Immunoregulatory T Cells Suppress Polyclonal T Cell Activation In Vitro by Inhibiting Interleukin 2 Production. J Exp Med.1998; 188: 287-296.
- Gravano DM, Vignali DA. The Battle Against Immunopathology: Infectious Tolerance Mediated by Regulatory T Cells. Cell Mol Life Sci. 2012; 69: 1997-2008.
- Jonuleit H, Schmitt E, Kakirman H, Stassen M, Knop J, Enk AH. Infectious Tolerance: Human CD25(+) Regulatory T Cells Convey Suppressor Activity to Conventional CD4(+) T Helper Cells. J Exp Med. 2002; 196: 255-260.
- Jonuleit H, Schmitt E. The Regulatory T Cell Family: Distinct Subsets and their Interrelations. J Immunol. 2003; 171: 6323-6237.
- Zhang S, Wu K, Liu Y, Lin Y, Zhang X, Zhou J, et al. Finasteride Enhances the Generation of Human Myeloid-Derived Suppressor Cells by Up-Regulating the COX2/PGE(2) Pathway. PLoS One. 2016; 11: e0156549.
- Lim J-Y, Park M-J, Im K-I, Kim N, Jeon E-J, Kim E-J, et al. Combination Cell Therapy Using Mesenchymal Stem Cells and Regulatory T cells Provides a Synergistic Immunomodulatory Effect Associated With Reciprocal Regulation of Th1/Th2 and Th17/Treg Cells in a Murine Acute Graft-Versus-Host Disease Model. Cell Transplant. 2014; 23: 703-714.
Citation:Siegmund DM, Schäfer I, Koch R, Singh A, Handgretinger R, Rieber N, et al. Functional Assessment of Myeloid-Derived Suppressor Cells, Mesenchymal Stromal Cells, and Regulatory T Cells for the Control of T Cell Function: Implications for Graft-versus-Host Disease. Ann Hematol Oncol. 2017; 4(1): 1132. ISSN : 2375-7965