
Case Report
Ann Hematol Oncol. 2017; 4(6): 1156.
Cardiac Primitive Neuroectodermal Tumor (PNET) with Metastasis at Presentation
Jawahar A*, Hibbeln J and Goldberg A
Department of Radiology, Loyola University Medical Center, USA
*Corresponding author: Jawahar A, Department of Radiology, Loyola University Medical Center, 2160, South 1st Avenue, Maywood, Illinois-60153, USA
Received: April 25, 2017; Accepted: May 31, 2017; Published: June 22, 2017
Abstract
Primitive neuroectodermal tumors (PNET) are rare tumors and PNET of cardiac origin are extremely rare, with only 6 cases reported since 1996. Cardiac PNET cases have been associated with poor prognosis with extensive metastasis at presentation, and warrant aggressive treatment.
We present a 19-year-old male patient who presented with headache and falls; workup demonstrated cerebellar lesions which were resected and noted to be metastatic PNET, and his preoperative transthoracic echocardiography showed an irregular cardiac mass protruding into the interventricular septum which was later identified as the primary malignant mass. Despite the mass being of the cardiac primary origin, he did not have cardiac symptoms until 15 months after his initial symptoms.
Keywords: PNET; Cardiac tumors; Metastasis; Neuroectodermal tumors; Cardiac transplantation
Introduction
Cardiac PNET is an extremely rare tumor, with only 6 cases reported in literature to date since it was first described by Charney, et al. in 1996 [1]. Cardiac PNETs have poor prognosis with extensive metastasis at presentation, and have been felt to warrant aggressive treatment with chemotherapy and radiotherapy, with limited surgical options [2-4]. Though recurrences have been commonly reported, there have been two cases described in which complete resection of tumor in patients without extracardiac or metastatic spread was shown to be beneficial, without recurrence in subsequent short follow up. The role of imaging is to identify the primary tumor, its invasion into its surrounding structures, and metastatic foci which can make these patients non-surgical candidates, more appropriately treated with chemotherapy and radiotherapy.
Case Presentation
A 19-year-old male presented to our institution in November 2015 with complaints of worsening headache for 2 months and subsequent imbalance with falls. A CT head (Figure 1A,B) showed three ill-defined cerebellar lesions suspicious for malignancy and underwent craniotomy with excision of the masses with negative margins. Immunohistochemical stains of the lesions were positive for cytokeratin CAN 5.2, GFAP, vimentin and CD56, and negative for translocation characteristic of synovial sarcoma. Histopathology was reported as metastatic primitive neuroectodermal tumor and he received post-operative radiotherapy for 3 weeks. Preoperative transthoracic echocardiography (Figure 2A) showed an approximately 5 cm irregular mass involving and protruding from the interventricular septum, with involvement of both ventricles. The uninvolved myocardium contracted normally; left ventricular ejection was 55%. Cardiac MRI (Figure 2B, 2C, 2D) confirmed a 5.2x4.9 cm irregular mass infiltrating the mid interventricular septum with extension into both ventricular cavities. The mass was isointense on T1W images, hyperintense on T2W images, and showed robust perfusion on T1 post-contrast imaging. Delayed hyperenhancement was also seen. No obvious obstruction of cardiac blood flow was noted.
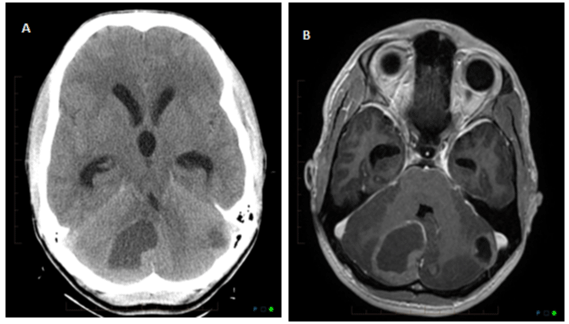
Figure 1: Unenhanced axial CT head (1A) and axial T1 fat saturated post
contrast (1B) images show ill-defined hypodense lesions in right and left
cerebellar hemispheres, larger one in the right cerebellar hemisphere. There
is mass effect with compression of the fourth ventricle and mild dilatation of
third and lateral ventricles.
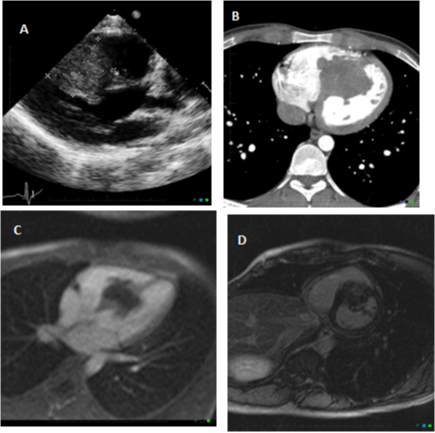
Figure 2: Transthoracic echocardiography (2A) and contrast-enhanced
axial cardiac CT chest (2B) showed a lobulated mass arising from
the interventricular septum and extending into the both ventricular
chambers without outflow obstruction. Cardiac MRI pre- and post-contrast
administration (2C,2D) showed a 5.2x4.9 cm irregular mass infiltrating the
mid interventricular septum with extension into both ventricular cavities. The
mass was isointense on T1W images, hyperintense on T2W images, and
showed robust perfusion on T1 post-contrast imaging.
Delayed hyperenhancement was also seen.
CT chest, abdomen and pelvis (Figure 3A) for evaluation of a palpable right flank mass showed hypo-enhancing soft tissue mass measuring 4.9x2.6 cm in the subcutaneous plane of the right posterolateral abdominal wall above the iliac crest. He was also noted to have multiple pulmonary nodules, with the largest nodule measuring 1.2 cm. Incisional biopsy of the right flank mass was reported as a malignant small round cell neoplasm favoring desmoplastic small round cell tumor (Figure 4) with tumor cells positive for cytokeratins, AE1/AE3, vimentin, focally bcl2 and NSE; negative for S100, CD34, synaptophysin, chromogranin and desmin, and negative neuroendocrine markers. The pathology slide was sent to NIH/NCI and was reported as malignant small round cell tumor. FISH analysis was positive for EWSR1 gene rearrangement at 22q12 locus that is commonly associated with Ewing sarcoma or PNET. He underwent bone marrow biopsy which showed hypocellular bone marrow for age (40%-50% cellular) with negative metastatic tumor in the bone marrow. Radical resection of the cardiac mass was deferred due to his uncontrolled disseminated disease and poor prognosis. Repeat imaging 6-months from initial presentation showed new rimenhancing lesions in the bilateral frontal lobes which were suspicious for metastatic lesions without residual or recurrent tumor at the postoperative site in cerebellum (Figure 3B). He was irradiated for his brain lesions which showed substantial response to radiation. He was started on chemotherapy as per MSKCC regimen of standard chemotherapy, with irinotecan, temozolomide, and bevacizumab. During his chemotherapy treatment, CT chest, abdomen and pelvis showed increase in the right flank subcutaneous mass with new and enlarging pulmonary nodules, new retroperitoneal nodule, and retrocrural and inguinal lymphadenopathy, demonstrating progression of metastatic disease; however, the cardiac mass remained grossly stable to mildly increased in size. He had several hospital admissions during the chemotherapy treatment for side effects of neutropenic fever, epistaxis from thrombocytopenia. During his 7th cycle of chemotherapy regimen in January 2017, he had an episode of tonic-clonic seizures, followed by hypotension and cardiac arrest from ventricular fibrillation. ACLS algorithm was initiated, intubated and there was return of spontaneous circulation after 15-20 minutes. He was subsequently made DNR/DNI, comfort care was continued and discharged to home hospice.
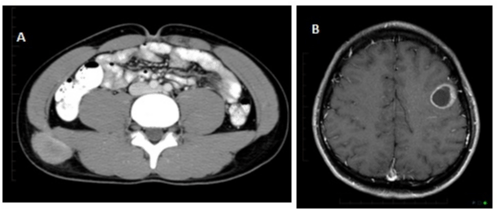
Figure 3: CT abdomen and pelvis (3A) performed for metastatic workup
showed an irregular, mildly enhancing 2.6 x 4.9 cm soft tissue mass with small
adjacent nodule in the posterior lateral abdominal wall immediately above the
right iliac crest. MRI brain- axial T1 fat-saturated post contrast image showed
a rim-enhancing lesion in the left posterior frontal lobe measuring 2.5x2.2 cm
with minimal surrounding vasogenic edema, consistent with new metastatic
lesion (3B).
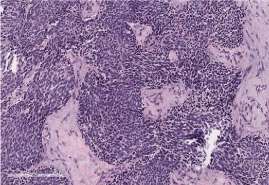
Figure 4: Histopathology shows proliferation of round variably sized and
shaped cells forming nests and surrounded by prominent desmoplastic
stroma.
Discussion
The natural history of cardiac tumors has been revolutionized by the prevalent use of echocardiography and advanced surgical techniques ranging from simple resection of intrapericardial tumors to cardiac transplantation for unresectable tumors with the aid of extracorporeal circulation [5]. Cardiac tumors are metastatic at presentation in 80% of cases and primary cardiac tumors constitute only 0.001%-0.28% of autopsy series [2,3,6]. Among the common primary tumors, atrial myxoma constitutes 70%-77% [5,7]. The most common primary malignant cardiac tumors include sarcomas, followed by lymphoma and mesothelioma [2,8]. PNET involving the myocardium or epicardium is extremely rare; however, it is the most common type of sarcoma in the first 2 decades of life [9].
The first myocardial PNET case was reported in 1996 by Charney, et al. [1,10] primary cardiac PNETs have been reported only 6 times in the literature until 2012 [1,2,4,9-11]. One case of metastatic PNET to the heart has been described by Naseri, et al. in 2005 [12]. PNETs in general are rare, highly malignant, small round cell tumors affecting bone and soft tissues and carry very poor prognosis in adults [13]. Histopathology shows classical small round blue cells of undifferentiated neuroectodermal origin [13]. PNET was first described as a tumor of neural origin by Stout in 1935, however it was later classified in 1996 by Batsakis, et al. into central, autonomic, and peripheral PNETs [13,14]. PNET can be differentiated from Ewing sarcoma by its neurosecretory granules on electron microscopic examination [14]. The antigen patterns in staining for neuronspecific enolase (NSE) and periodic acid-Schiff place the PNET histopathologically between Ewing sarcoma and neuroblastoma [14]. Criteria for morphological diagnosis of malignant PNET include positive NSE immunochemical staining, pseudorosette formation on light microscopy, neurosecretory granules on electron microscopy, and presence of a balanced reciprocal translocation t(11;22) causing alteration of the pleuripotent neural crest cells [9].
Clinical presentation of cardiac PNET is dependent on the involvement of valves, myocardium or pericardium by the malignant mass [2]. Hence, clinical features have been reported to include myocardial ischemia, congestive right or left heart failure, arrhythmias, thromboembolism, pericardial effusion, cardiac tamponade, or invasion of adjacent mediastinal structures [2,9,10- 12,15]. Symptoms can also include weight loss, malaise, fatigue, exertional dyspnea, and orthopnea [1,2,9,10]. Imaging evaluation is typically initiated by transthoracic echocardiography which can identify tumor location and growth pattern [2,5,10,12]. However, CT and MRI play an important role in the characterization of the mass and assessment of the extra-cardiac spread, especially in the presence of significant pericardial fluid which limits echocardiography utility [2,12,16]. Cardiac MRI, with and without gadolinium contrast, is useful to delineate the extent, viability, and vascular invasion of the mass, which is necessary to determine if the patient is a surgical candidate [12]. Imaging findings include pericardial effusion, heterogeneous myocardial or epicardial enhancing mass that can have cystic components, and often invasion of coronary arteries and/ or aorta [2,9,10]. It is difficult to arrive at a definitive diagnosis prior to surgery based on imaging features alone; hence endomyocardial biopsy or paracentesis pericardii is required for definitive diagnosis [9,10].
Microscopic examination of the resected mass or specimen is the gold standard examination as with any oncologic mass. Microscopic examination shows diffuse sheets of round cells separated by fibrous tissue with or without Homer-Wright pseudorosettes. These round cells show strong CD-99 membrane positivity, and positive neural markers including S-100 protein, neuron specific enolase (NSE), synaptophysin and chromogranin and negative staining for cytokeratin, actin, desmin, myoglobin, and leucocyte common antigen [1,2,10,15,16]. Characteristic cytogenetic abnormality is reciprocal translocations: t(11;22) or t(21; 22) [1,2,9]. Ewing’s sarcoma is difficult to differentiate from PNET due to similar anatomic locations, overlapping light microscopic appearance, cell biology, cytogenetics and wide age range at presentation [1]. However, Enzinger and Weiss’ criteria for the differentiation of the two include at least 2 neuronal markers, light microscopic rosettes, or ultrastructural evidence of neural differentiation for diagnosis of PNET over extraosseous Ewing’s sarcoma. Our patient had cardiac and bilateral cerebellar lesions at initial presentation. The symptomatic cerebellar lesion was excised which was reported as metastatic PNET on histopathology. Also, there has been no reported case in literature describing cerebellar PNET metastatic to heart. Hence, the larger cardiac mass with multifocal smaller brain lesions are consistent with primary cardiac tumor in our patient. The flank mass and pulmonary nodules were metastatic by chronology of development and also with the flank mass being biopsy-proven to be metastasis.
In general, aggressive cardiac tumors are often treated surgical due to the high preponderance of local recurrence, along with pre-surgical/postsurgical chemotherapy and/or radiotherapy [14]. Orthotopic cardiac transplantation has been performed for unresectable locally aggressive cardiac tumors without extracardiac involvement, however this has not been shown to be superior to conventional treatment of chemotherapy and radiotherapy [3- 5,7,10,12]. Cardiac PNET, in particular, has poor prognosis due to its infiltrative and invasive nature and, therefore, chemotherapy and radiotherapy are the mainstay of treatment with limited surgical options [2-4]. Rajappa, et al. described that chemotherapy caused 50% reduction in the size of the mass in their patient with symptomatic relief and reversal of EKG changes [2]. This is consistent with experience with other PNET tumors, for which chemotherapy and radiotherapy have been utilized to achieve partial remission prior to surgery [9]. The P6 chemotherapy protocol used for our patient was based on the Memorial Sloan Kettering protocol which is a high-dose alkylator-based regimen for desmoplastic small round cell tumors and had shown efficacy in poor prognostic tumors, with relatively non-toxic nature and non-cross-resistance to the regimen [17].
Recurrence with PNETs is usually locally aggressive and extensive surgery is required [10,14]. In a large trial of 42 patients with malignant PNET by Jurgens, et al. in 1987, he found that there was striking tendency for local recurrence which emphasized the importance of significant surgery [14]. They also described metastases were more common to bone and lungs, and most patients received combined modality treatment with extensive surgery, chemotherapy, and radiation therapy [14]. Kusher, et al. in their experience at Memorial Sloan-Kettering center described 25% survival rate in patients with tumor larger than 5 cm [1]. Long term cardiac PNET prognosis has not been adequately described due to the small numbers of cases.
In conclusion, cardiac PNETs are extremely rare tumors, typically with extracardiac manifestations at initial presentation, often rendering limited surgical options for these patients. Imaging plays a crucial role in diagnosis, guiding therapy by identifying surgical candidates versus non-surgical treatment, and in follow up of patients.
References
- Charney DA, Charney JM, Ghali V, Teplitz C. Primitive neuroectodermal tumor of the myocardium: A case report, review of the literature, immunohistochemical and ultrastructural study. Hum Pathol. 1996; 27: 1365- 1369.
- Rajappa S, Gundeti S, Varadpande L, Bethune N, Rao S, DigumartiR. Cardiac Primitive Neuroectodermal Tumor Presenting As Acute Coronary Syndrome. J Clin Oncol. 2007; 25: 449-451.
- Mazuecos JJ, Manso RF, Cubero JS, Ramos JT, Dominguez JO, Rivera LA. Is heart transplantation for primary cardiac sarcoma a useful therapeutic option? Rev Esp Cardiol. 2003; 56: 408-411.
- Thachil A, Saxena A, Choudhary UK, Ray R. Primary primitive neuroectodermal tumor: An unusal case of right ventricular intracavitary obstruction in a child. Ann Pediatr Cardiol. 2008; 1: 147-148.
- Basso C, Valente M, Poletti A, Casarotto D, Thiene G. Surgical pathology of primary cardiac and pericardial tumors1. Eur J Cardiothorac Surg. 1997; 12: 730-738.
- Abraham KP1, Reddy V, Gattuso P. Neoplasms metastatic to the heart: review of 3314 consecutive autopsies. Am J Cardiovasc Pathol. 1990; 3: 195-198.
- Besirli K, Arslan C, Tuzun H, Oz B. The primitive neuroectodermal tumor of the heart. Eur J Cardio-thorac Surg. 2000; 18: 619-621.
- Chai Y, Huang L, Yue L. Peripheral primitive neuroectodermal tumor of left ventricular wall origin: a rare case. Acta Cardiol. 2007; 62: 523-524.
- Purkayastha A, Pathak A, Sharma N, Viswanath S, Dutta V. Primitive neuroectodermal tumor of lungs in adults: a rare series of three cases treated with upfront chemo-radiation. Transl Lung Cancer Res. 2016; 5: 350-355.
- Jürgens H1, Bier V, Harms D, Beck J, Brandeis W, Etspüler G, et al. Malignant peripheral neuroectodermal tumors. A retrospective analysis of 42 patients. Cancer. 1988; 61: 349-357.
- Goldstein DJ, Oz MC, Rose EA, Fisher P, Michler RE. Experience with heart transplantation for cardiac tumors. J Heart Lung Transplant. 1995; 14: 382- 386.
- Perchinsnky MJ, Lichtenstein SV, Tyers GF. Primary cardiac tumors: Forty years’ experience with 71 patients. Cancer. 1997; 79: 1809-1815.
- Beşirli K1, Arslan C, Tüzün H, Oz B. The primitive neuroectodermal tumor of the heart. Eur J Cardiothorac Surg. 2000; 18: 619-621.
- Araoz PA1, Eklund HE, Welch TJ, Breen JF. CT and MR imaging of primary cardiac malignancies. Radiographics. 1999; 19: 1421-1434.
- Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, et al. Desmoplastic Small Round-Cell Tumor: Prolonged Progression-Free Survival With Aggressive Multimodality Therapy. J Clin Oncol. 1996; 14: 1526-1531.
- Naseri E, Eralp B, Oztek I. Emergency management of severe right ventricular inflow obstruction secondary to a metastatic cardiac tumor. Ann Thorac Surg. 2005; 79: 709-711.
- Nwaejike N, Rassl D, Ford H, Large SR. Primitive neuroectodermal tumor of the heart. Ann Thorac Surg. 2012; 93: e27-29.