
Case Report
Ann Hematol Oncol. 2017; 4(10): 1176.
ARC Syndrome and Platelet’s Abnormalities about Two Cases
Maaloul I¹*, Chabchoub I¹, Chaari M², Jribi S², Elleuch H², Aloulou H¹ and Hachicha M¹
¹Department of Pediatrics, Hedi Chaker Hospital, Tunisia
²Laboratory of Hematology, Hedi Chaker Hospital, Tunisia
*Corresponding author: Maaloul I, Department of Pedicatrics, Hedi Chaker Hospital, El Ain Street, Tunisia
Received: May 08, 2017; Accepted: November 07, 2017; Published: November 15, 2017
Abstract
Introduction: ARC syndrome (arthrogryposis-renal dysfunctioncholestasis) is a rare lethal multisystemic autosomal recessive disease due to a mutation in the VP33B gene on chromosome 15q26.1.
Case Presentation: The authors reported two newborns of Tunisian consanguineous parents, who presented the three characteristic features of ARC syndrome. Their blood smear showed large, pale platelets which is characteristic in this syndrome. The two children were dead respectively at the age of 40 days and 2.5 months because of sepsis.
Conclusion: The presence of granular and large platelets, in newborns suffering from cholestasis, proximal tubulopathy and orthopedic problems should be considered as an important diagnosis criteria.
Keywords: Arthrogryposis-renal dysfunction- cholestasis syndrome; Cholestasis; Blood smear; Gray platelets; Child
Abbreviations
ARC: Arthrogryposis Renal Dysfunction Cholestasis; g-GT: gamma-Glutamyltranspeptidase; GGT: Gamma Glutamyl Transferase
Introduction
Since the first report of arthrogryposis renal dysfunction and cholestasis (ARC) syndrome by Lutz-Richter and Landolt in 1973 [1], this very rare and lethal infantile disease has been reported worldwide. ARC syndrome is a rare multisystem disorder which is fatal; death occurs generally in the first year of life. It’s an autosomal recessive disease due to a mutation in the VP33B gene on chromosome 15q26.1 [2]. The cardinal clinical features of the syndrome include cholestasis with normal serum gamma-glutamyltranspeptidase (g-GT) activity, arthrogryposis, and renal disorders (renal failure, renal tubular dysfunction) [1-3]. Bleeding problems have also been observed in this syndrome secondary to abnormal platelet’s morphology and function. Few reports on ARC syndrome have described abnormal platelets [2,4-6]. We report two new cases of ACR syndrome who presented documented abnormality of platelet’s morphology and function.
Cases Presentation
Case 1
A male infant was admitted at the age of 9 days for jaundice which was noticed on day 5. He was the fifth child to consanguineous healthy parents. He was born at term after an uneventful pregnancy, weighting 3300 g (50th percentile). There was a history of death of two brothers respectively at the age of 40 days and 4 months; they presented neonatal jaundice and skeletal abnormalities, whereas, his two sisters were healthy. At birth, arthrogryposis was noticed, but no evident other malformations. On admission, the clinical examination revealed dysmophism (low set ears, arched palate and retrognatism), ichthyosis, arthrogryposis (Figure 1), jaundice with normal colored stools, but no hepatomegaly. Laboratory tests showed high total bilirubin (366.6 μmol/l) and direct bilirubin (247 μmol/l), whereas, alanine transferase and aspartate aminotransferase levels were normal. Gamma-glutamyl–transferase (GGT) activity was normal (21 U/l). Biological tests revealed hyperchloremic metabolic acidosis with mild proteinuria and increased urea (14.2 mmol/l) and creatinine (92 μmol/l). Thyroid tests functions were normal and urine culture was negative for bacteria.
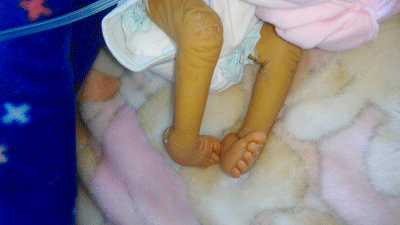
Figure 1: Patient no1: arhrogryposis, ichtyosis.
Abdominal ultrasound examination was normal.
After initial investigations, he was discharged and was readmitted at 30 days of life when he presented a mild dehydration and growth failure. His weight was 2.6 kg (-2,5DS), his height was 49 cm (-2DS). We noticed a bleeding tendency during blood sampling. The platelet count was normal, however platelet morphology was abnormal. In fact, the blood smear showed pale-appearing platelets consistent with gray platelet syndrome (Figure 2). Platelet Function Analyser (PFA100) was prolonged. The patient showed marked growth failure despite of high caloric enteral feeding and presented two febrile episodes. He was dead at the age of 40 days because of severe sepsis.

Figure 2: Blood smear of patient no1: large and pale platelets.
Case 2
A male infant, born to consanguineous parents, was admitted at the age of 40 days, because of jaundice noticed since 5 days of life. His brother was died at the age of 50 days because of icterus and bacterial sepsis. The infant was the fourth child of the couple, born at term, weighting 3400 g. The clinical examination revealed dysmorphic features, hypotonia and icterus without hepatomegaly, partial discolored stools and club feet. Liver investigations were performed which revealed hyperbilirubinemia, normal transaminases, and normal gamma glutamyltranspeptidase (AST: 57, ALT: 45; Total bilirubin: 175.2 μmol/l, direct Bilirubin: 161 μmol/l; GGT: 12UI/l). Prothrombin time was normal (91%). He had hyperchloremic metabolic acidosis without glucosuria, normal urea and creatinine thyroid function tests were normal. Abdominal ultrasound was normal.
Complete blood count showed severe anemia (Hb 5.9 g/dl) and normal platelet count. He had prolonged PFA 100. Peripheral blood smear revealed large and pale platelets (Figure 3). Despite continuous enteric feeding, the infant had marked failure to thrive and presented several episodes of dehydration with hypernatremia despite the absence of gastrointestinal troubles, these troubles were explained by polyuria.
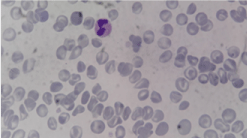
Figure 3: Blood smear of the patient no2 showing abnormal morphology of
the platelets: large and pale.
Moreover, he had two episodes of bacterial sepsis. The first was due to staphylococcus aureus that was treated by intravenous antibiotics. The second was due to Klebsiella pneumonia multiresistant that resulted in his death at the age of 2.5 months.
Discussion
1. Most of the reported cases of ACR syndrome in the literature were in Pakistani infants, rarely in Saudi Arabia, Turkey, Oman, North Africa , Italy , Asia and Portugal [5] ; suggesting that the occurrence of this condition is common in communities where the rate of consanguineous marriage is very high. Our patients were born to consanguineous parents and have a charged family’s history of similar cases.
2. ACR syndrome is a fatal condition characterized by three key features: arthrogryposis, renal tubular dysfunction and cholestasis with low or normal GGT levels [2,3]; this trial was the basis of the diagnosis of ARC syndrome in our patients , because molecular investigations of this syndrome are currently unavailable in our country. Other clinical manifestations were observed, including dysmorphic features, failure to thrive, ichtyosis, hypothyroidism, and congenital heart disease, cerebral malformations like agenesis of the corpus callosum, nerve deafness, recurrent infections and platelet abnormalities [3].
3. Structural abnormalities seen in ARC platelets are similar to those observed in gray platelet syndrome: increased platelet size, a pale appearance in blood films, and absence of alpha granules with increased γ granules [7]. Platelet function is affected; most authors observed reduced aggregation with arachidonate and adenosine diphosphate (ADP) [4,7], they conclude that VPS33B is included in intracellular vesicle trafficking, being essential for the development of platelet α granules [7].
This hematological manifestation was considered by Kim and al as a cardinal feature of this condition and can be useful as a non-invasive diagnostic marker for ARC syndrome [4]. In fact, they reported 12 cases of ACR syndrome; in all cases, close examination of peripheral blood smear showed large, pale, granular platelets similar to those seen in gray platelet syndrome. Thus, peripheral smear can be used as it is available and non-invasive to screen for bleeding disorders in patients with ARC syndrome. Because of platelets abnormalities in ARC syndrome, invasive procedure such us organ biopsy may cause bleeding and should be avoided.
Our patients had large, pale appearance platelet in addition to the characteristic features of the syndrome.
4. As a lethal multisystem disorder, prognosis in ARC disorder is very poor. Most patients succumb within the first year of life after developing recurrent infections, severe hydropenia du to tubular dysfunction, acidosis or internal hemorrhaging [3].
Conclusion
The combination of cholestasis, tubular disorder (acidosis, polyuria) and orthopedic problems suggests the diagnosis of ARC syndrome. A blood peripheral smear greatly facilitates the diagnosis in countries where the molecular investigation is not available. Unfortunately, prenatal diagnosis cannot be performed to prevent the recurrence of such lethal disease in the following pregnancies.
References
- Lutz-Richner AR, Landolt RF. Familiaregallengangsmissbil-dungenmittubular erneireninsurfizienz. Helv Paediatr Acta. 1973; 28: 1-12.
- Giraud A, Ramond F, Cremillieux C, Touraine R, Patural H, Stephan JL. The complex phenotype of ARC syndrome: A new case. Arch Pediatr. 2016; 24: 131-134.
- Zhou Y, Zhang J. Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: from molecular genetics to clinical features. Ital J Pediatr. 2014; 40: 77.
- Kim SM, Chang HK, Song JW, Koh H, Han SJ. Agranular platelets as a cardinal feature of ARC synrdrome. J Pediatr Hematol Oncol. 2010; 32: 253- 258.
- Saadah O, Bokhari B, Alshaeri T, Jastaniah W. Haematological manifestations of arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: a case report. Arab J Gastroenterol. 2013; 14: 26-28.
- Ilhan O, Ozer EA, Ozdemir SA, Akbay S, Memur S, Kanar B, et al. Arthrogryposis-renal tubular dysfunction-cholestasis syndrome: a cause of neonatal cholestasis. Case report. Arch Argent Pediatr. 2016; 114: e9-e12.
- Lo B, Li L, Gissen P, Christensen H, McKiernan PJ, Ye C, et al. Requirement of VPS33B, a member of the Sec1/Munc 18 protein family, in megakaryocyte and platelet α-granule biogenesis. Blood. 2005; 106: 4159-4166.