
Case Report
Ann Hematol Oncol. 2018; 5(4): 1205.
Successful Treatment of Macrophage Activation Syndrome and Symptom of Inappropriate Anti- Diuretic Hormone in Chronic Natural Killer Cell Lymphoproliferative Disorder with Cyclophosphamide
Sarkissian S1*, Siaghai PJ2, Mkhikian H2, Jeyakuma D1 and Rezk S2
1Department of Hematology and Oncology, University of California, USA
2Department of Pathology, University of California, USA
*Corresponding author: Sarkissian S, Department of Hematology and Oncology, University of California at Irvine, USA
Received: May 03, 2018; Accepted: May 30, 2018; Published: June 12, 2018
Abstract
Chronic natural killer (NK) cell lympho proliferative disorders (LPD) are very rare indolent hematologic disorder. Less than 100 cases have been reported world-wide. The disease is characterized by proliferation of NK cells, that have a CD3-, CD16+, CD56+ flow cytometry signature, and a non-rearranged T-cell receptor γ. Patients can present with cytopenias, macrophage activation syndrome and constitutional symptoms. Differentiation from malignant NK disorders such as aggressive natural killer cell leukemia’s and extra nodal NK/Tcell lymphoma, nasal type, is crucial.
We report the case of an elderly gentleman who presented with a several months history of leukopenia and constitutional symptoms. Diagnostic work-up revealed a Comb’s negative hemolytic anemia, SIADH, macrophage activation syndrome, hyper cellular bone marrow, splenomegaly and extra-nodal disease. His flow cytometry signatures showed a unique CD2+ CD3- CD7+ CD16+ and CD56dim lymphocyte population, which comprised 16% of the total lymphocyte population. T-cell receptor gene γ rearrangement as well as B-cell markers was negative. The patient was refractory to corticosteroid and IVIG therapy. After several months of therapy with oral cyclophosphamide, he showed a robust response with improvement in his symptoms and resolution of his transfusion dependence
Chronic NKLPD is a rare disease and poses a diagnostic and treatment challenge for hematologists. The lack of a clonally marker and indolent nature of the disease make diagnosis very elusive and challenging. While some studies suggest clinical utility of unique molecular and flow cytometry features, the entity remains a diagnosis of exclusion. A lack of randomized clinical trials and biomarkers makes therapeutic decision-making contingent on short case series publications.
Keywords: Macrophage activation syndrome; Chronic natural killer; Cell lymphoproliferative disorder cyclophosphamide
Introduction
Large granular lymphocytes (LGL) are cytotoxic lymphocytes that play an important role in the innate immune system. Disorders of large granular lymphocytes can be either reactive to an underlying malignancy or autoimmune disease, or a primary clonal disorder. Reactive processes are polyclonal, while malignant processes are monoclonal. In the clonal disorder, the lymphocyte population has a T-cell receptor gene rearrangement in either the α/β orγ/δ configuration. Clonal T- LGL and reactive chronic LGL are defined by a chronic, 6-month, proliferation of LGL cells. T-cell LGL has a CD3+ CD4- CD8+ CD45- CD56± CD57+ phenotype. Chronic NKLPD, has a CD3- CD8+ CD16+ CD56+ CD57- phenotype. Natural killer cell neoplasms lack the T-cell receptor gene rearrangement. The distinction between the 2 entities can be made via flow cytometry and T-cell receptor gene rearrangement. In the setting of an un-rearranged T-cell receptor, diagnosis is made via exclusion [1-11].
In 2008, the World Health Organization, WHO, distinguished 3 different LGL disorders, T-cell LGL, Chronic NKLPD, aggressive NK/T cell leukemia/lymphoma (ANKL) (of which, extra-nodal NK/Tcell leukemia/lymphoma, nasal type, is an entity). Clinical distinction between chronic NKLPD and T-cell LGL can be challenging as they have relatively similar clinical presentation. Mutations in the STAT3 gene have been shown to be a common marker of pathogenesis. While there is no universally validated clonal marker, CD94 (hi)/HLADR+ lymphocyte positivity and lack of Killer Cell Immunoglobulin Receptor (KIR3DL1) expression have been proposed. Given the rarity of chronic natural killer cell lymphoproliferative disorder, only a few case series have been published. The largest registry study, published by the French, showed the following: The presence of constitutional symptoms (30%), autoimmune disease (24%, splenomegaly (14%), recurrent infections (14%), neuropathy (3%). 13% and 11% had a reported prior solid malignancy or hematologic malignancy, respectively. 15% had an autoimmune cytopenia and 51% had a reported neutropenia [12-25].
Given the rarity of Chronic NK cell Lymphoproliferative Disorder, there are no randomized trials helping direct the ideal mode of therapy. Much of the information has been extrapolated from treatment of LGL. Anecdotal data suggests that corticosteroids, cyclophosphamide, and methotrexate may prove to be efficacious. If there is a driver disease such as another neoplasm, infection, or autoimmune disease then the primary disease should be targeted as well. We report the case of a patient with unexplained constitutional symptoms, cytopenias, macrophage activation syndrome, and SIADH who we treated with cyclophosphamide [26-29].
Case Presentation
A 75 years old Caucasian gentleman, who was otherwise in good health, presented to his PCP with complaints of dyspnea on exertion and a dry cough. He was treated for bronchitis with a course of antibiotics with resolution of the cough but without improvement in his respiratory symptoms. Two months later, the patient had a syncopal episode, with work-up revealing a leukocytosis with 16,800 leukocytes (84% Lymphocytes), hemoglobin of 7.8 g/dL and platelets of 264. Further work-up revealed that he had a lactate dehydrogenize of 873 IU/L (91-180), haptoglobin 6.0 ng/mL (16-200), total bilirubin of 1.5 mg/dL (0.1 -1.0) and a negative Coomb’s test. Erythropoietin level was elevated at 76.7 ug/L. Peripheral smear showed numerous large atypical appearing lymphocytes with azurophilic granules suspicious for LGL (Figure 1,2 and 3). There was a high suspicion for macrophage activation syndrome given his Ferritin of 7,358 ng/ mL, triglycerides of 290 mg/dL (‹150) and an elevated Interleukin-2 receptor of 7,250 pg/mL (‹1,032 pg/mL). Viral studies were negative for CMV, EBV and HHV-8. Computed tomography of the abdomen showed a 14 cm spleen, as well as numerous thoracic spine lesions. He underwent a bone marrow biopsy, revealing a hyper cellular marrow with appropriate trilineage hematopoiesis, erythroid hyperplasia and increased natural killer cell-like cells (Figure 4,5 and 6). There was no evidence of active hemophagocytosis on the bone marrow biopsy. Flow Cytometry showed that 70% of the lymphocytes were CD2+, CD7+, CD16+ while negative for CD 3,5,4,8, and 57 (Figure 2 and 3).
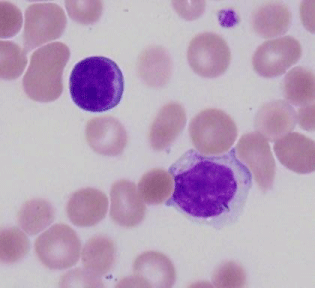
Figure 1: Peripheral blood smear showed increased circulating mature lymphocytes characterized by round nuclear contours, condensed chromatin, and moderate pale gray cytoplasm containing variably distinct azurophilic granules characteristic of large granular lymphocytes. Normal lymphocytes upper left for comparison.
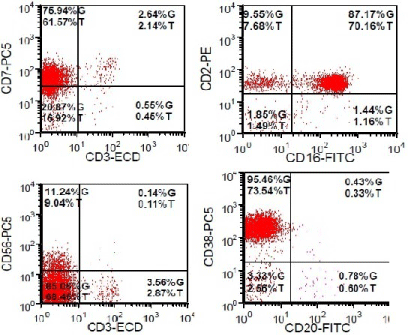
Figure 2: The majority of the lymphocytes are NK/NK-like cells with expression of CD 2, CD 7, CD 16, CD 38, and CD 56 (subset). They are negative for CD 3, CD 4, CD 8, and CD 5.
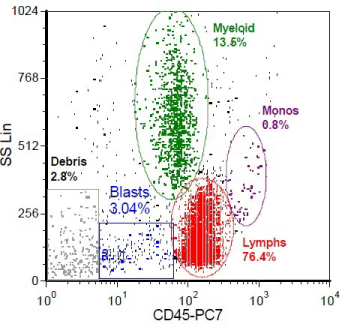
Figure 3: Flow cytometry of peripheral blood shows a population with bright CD 45 and low side scatter consistent with lymphocytes..
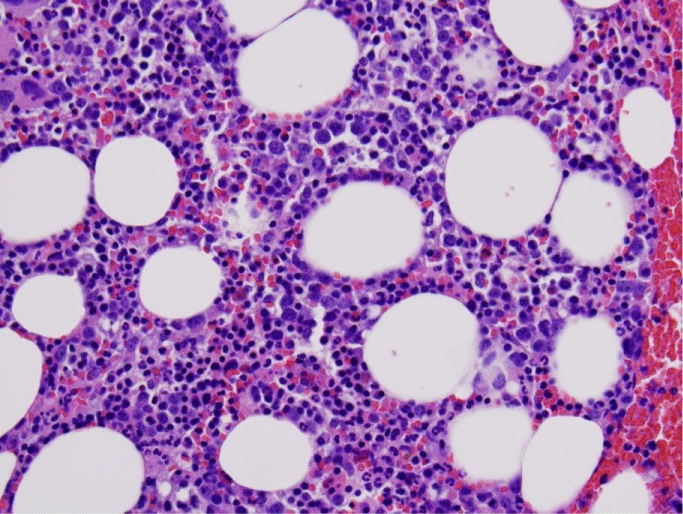
Figure 4: Bone marrow biopsy with a subtle interstitial and intrasinusoidal lymphoid infiltrate, best seen by immunohistochemical stains.).
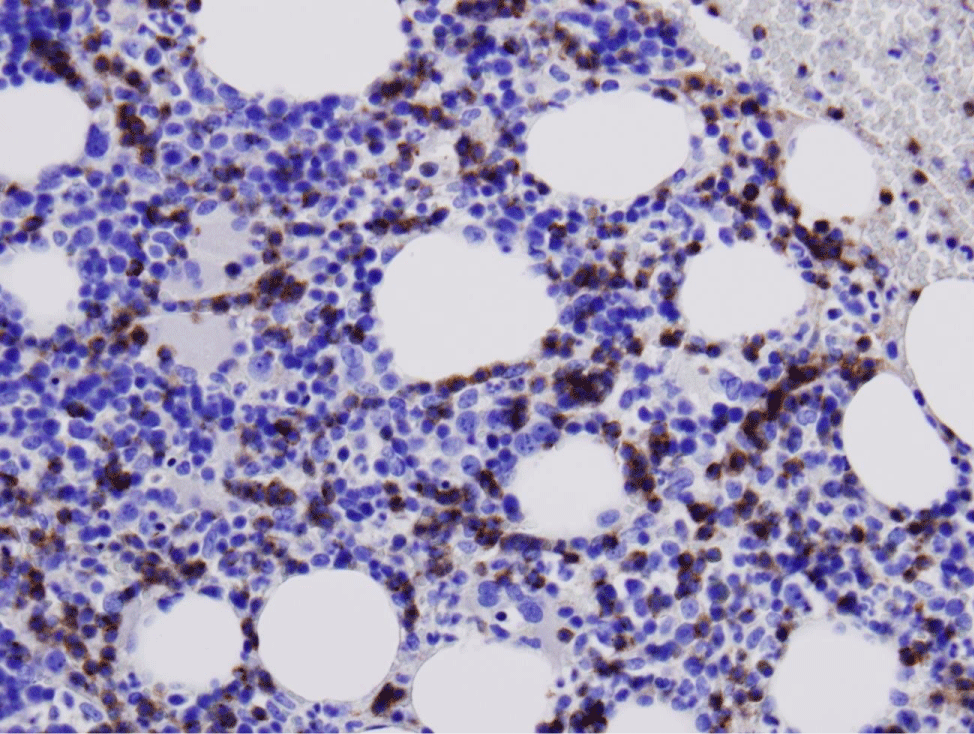
Figure 5: Increased CD2 staining highlighting the interstitial and intrasinusoidal lymphoid infiltrate.
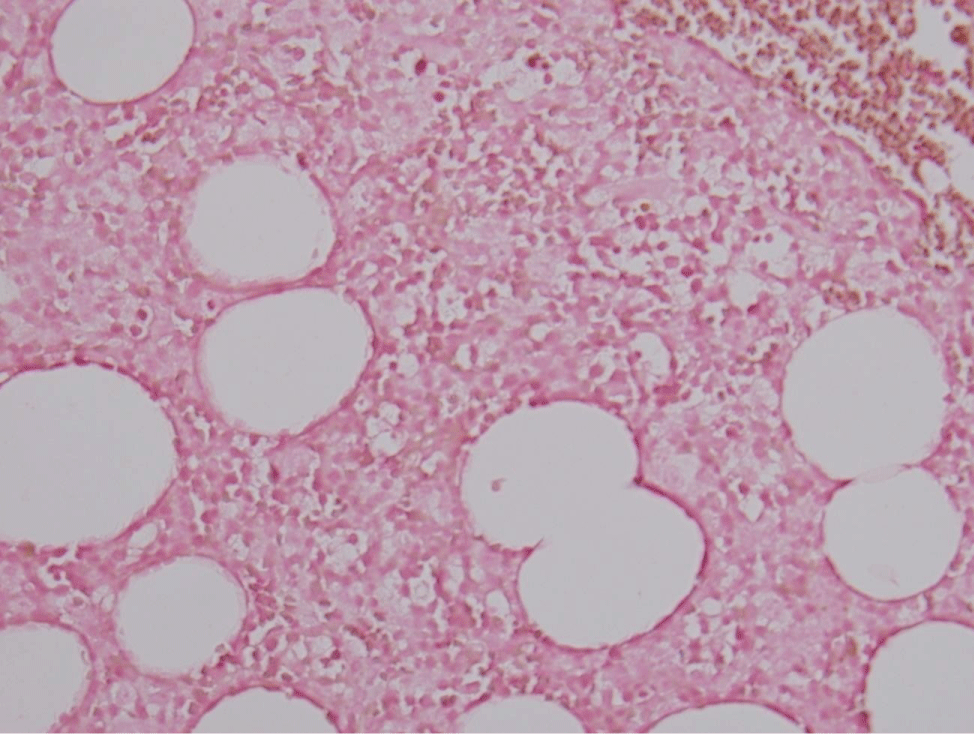
Figure 6: Negative ISH-EBV on bone marrow core.
He was started on treatment with prednisone 60mg daily as well as two infusions of IVIG 1 g/kg/day. The patient remained red blood cell transfusion dependent, requiring numerous units over the course of several weeks. After one month of prednisone therapy, his clinical condition continued to deteriorate with recurrent fevers, night sweats, mentation changes, and hyponatremia. Sepsis was ruled out as a cause of his fevers, and it was thought to be secondary to his underlying MAS. His sodium level was 123 mEq/L (135-145). Work-up of his hyponatremia was consistent with SIADH. This was thought to be a paraneoplastic phenomenon arising from his NK cell LPD. He was started on oral cyclophosphamide 50 mg daily. After several weeks of treatment, his sodium level corrected to 135 mEq/L, as did his mentation. His cyclophosphamide was then increased to 100mg daily and maintained at that dose. After several weeks, his constitutional symptoms resolved as did his transfusion dependence. After one year of therapy, his most recent labs showed a leukocyte count of 3,700, with 2,100 lymphocytes, hemoglobin 13.4 g/dL, and LDH 196 IU/L.
Discussion
Natural killer cell neoplasms remain a diagnostic challenge for many hematologists. In the background of another chronic disease process, they can be either a reactive or primary malignant process. The lack of a uniform and validated clonal marker has made the diagnosis challenging. Given its rarity in the Western hemisphere, experience is rather limited compared to Epstein-Barr virus endemic regions, such as central America and the Asian countries. Important variables to consider are the indolent nature of the disease, the presence of potential driver conditions, T-cell receptor clonality, autoimmune features, constitutional symptoms, and the flow cytometry profile. The pathogenesis appears to have some overlap with LGL, as described above, subtle diagnostic and clinical features may help distinguish the two. While CD94 (hi)/HLADR+ positivity and lack of Killer Cell Immunoglobulin Receptor (KIR3DL1), have been proposed, they have not been validated. There is also some contention as to the distinction between a common clone for Chronic NKLPD versus aggressive NK cell leukemia, and extra-nodal NK cell leukemia/lymphoma, nasal type. Some argue the more aggressive variants arise from the indolent disease. The rarity of this disease cohort makes treatment consensus challenging. Case reports and Case series can help clinicians guide therapy. Further research into the pathogenesis of this disease is crucial in developing efficacious therapeutics.
References
- Lamy T, Loughran TP Jr. Clinical features of large granular lymphocyte leukemia. Semin Hematol. 2003; 40: 185-195.
- Bareau B, Rey J, Hamidou M, Donadieu J, Morcet J, Reman O, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica. 2010; 95: 1534-1541.
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood. 2017; 129: 1082-1094.
- Loughran TP Jr. Clonal diseases of large granular lymphocytes. Blood. 1993; 82: 1-14.
- Janka GE, Lehmberg K. Hemophagocytic syndromes--an update. Blood Rev. 2014; 28: 135-142.
- Jerez A, Clemente M, Makishima H, Koskela H, Leblanc F, Peng Ng K, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012; 120: 3048-3057.
- Rajala HL, Olson T, Clemente MJ, Lagström S, Ellonen P, Lundan T, et al; The analysis of clonal diversity and therapy responses using STAT3 mutations as a molecular marker in large granular lymphocytic leukemia. Haematologica. 2015; 100: 91-99.
- Osuji N, Matutes E, Tjonnfjord G, Grech H, Del Giudice I, Wotherspoon A, et al. T-cell large granular lymphocyte leukemia: A report on the treatment of 29 patients and a review of the literature. Cancer. 2006; 107: 570-578.
- Aribi a, Huh Y, Keating M, O’Brien S, Ferrajoli a, Faderl S, et al. T-cell large granular lymphocytic (T-LGL) leukemia: experience in a single institution over 8 years. Leuk Res. 2007; 31: 939-945.
- Oshimi K. Clinical Features, Pathogenesis, and Treatment of Large Granular Lymphocyte Leukemias. Intern Med. 2017; 56: 1759-1769.
- Zhang R, Shah MV, Loughran TP Jr. The root of many evils: indolent large granular lymphocyte leukaemia and associated disorders. Hematol Oncol. 2010; 28: 105-117.
- Morice G, Jevremovic D, Olteanu H, Roden A, Nowakowski G, Kroft S, et al. Chronic lymphoproliferative disorder of natural killer cells: a distinct entity with subtypes correlating with normal natural killer cell subsets; Leukemia. 2010; 24: 881-884.
- Bárcena P, Jara-Acevedo M, Tabernero MD, López A, Sánchez ML, García- Montero AC, et al. Phenotypic profile of expanded NK cells in chronic lymphoproliferative disorders: a surrogate marker for NK-cell clonality. Oncotarget. 2015; 6: 42938-42951.
- Lima M. Aggressive mature natural killer cell neoplasms: from epidemiology to diagnosis. Orphanet J Rare Dis. 2013; 8: 95.
- Gattazzo C, Teramo A, Miorin M, Scquizzato E, Cabrelle A, Balsamo M, et al. Lack of expression of inhibitory KIR3DL1 receptor in patients with natural killer cell-type lymphoproliferative disease of granular lymphocytes. Haematologica. 2010; 95: 1722-1729.
- Lima M, Almeida J, Montero AG et al. Clinicobiological, immunophenotypic, and molecular characteristics of monoclonal CD56-/+dim chronic natural killer cell. Am J Pathol. 2004; 165: 1117-1127.
- Tefferi A, Li CY, Witzig TE, Dhodapkar MV, Okuno SH, Phyliky RL. Chronic natural killer cell lymphocytosis: a descriptive clinical study. Blood. 1994; 84: 2721-2725.
- Tefferi A. Chronic natural killer cell lymphocytosis. Leuk Lymphoma. 1996; 20: 245-248.
- Rabbani GR, Phyliky RL, Tefferi A. A long-term study of patients with chronic natural killer cell lymphocytosis. Br J Haematol. 1999; 106: 960-966.
- Neben MA, Morice WG, Tefferi A. Clinical features in T-cell vs. natural killercell variants of large granular lymphocyte leukemia. Eur J Haematol. 2003; 71: 263-265.
- Villamor N, Morice WG, Chan WC. Chronic lymphoproliferative disorders of NK cells. In Swerdlow SH. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARD Lyon. 2008; 274-275.
- Pullot E, Zambelo R, Leblanc F Bareau B4, De March E2, Roussel M, et al. Chronic Natural Killer Cell Lymphoproliferative Disorders: Characteristics of an international cohort of 70 patietns. Annals of Oncology. 2014; 25: 2030- 2035
- Suzuki R, Suzumiya J, Nakamura S, Aoki S, Notoya A, Ozaki S, et al. Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia. 2004; 18: 763-770.
- Oshimi K, Kawa K, Nakamura S, Suzuki R, Suzumiya J, Yamaguchi M, et al. NK-cell neoplasms in Japan. Hematology. 2005; 10: 237-245.
- Oshimi K. Progress in understanding and managing natural killer-cell malignancies. Br J Haematol. 2007; 139: 532-544.
- Moignet A, Hasanali Z, Zambello R, Pavan L, Bareau B, Tournilhac O, et al. Cyclophosphamide as a first-line therapy in LGL leukemia. Leukemia. 2014; 28: 1134-1136.
- Sekiguchi N, Nishina S, Kawakami T, Sakai H, Senoo N, Senoo Y, et al. Oral cyclophosphamide was effective for Coombs-negative autoimmune hemolytic anemia in CD16+CD56- chronic lymphoproliferative disorder of NK-cells. Int J Hematology. 2017; 105: 854-85
- Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood. 2011; 117: 2764- 2774.
- Steinway SN, LeBlanc F, Loughran TP Jr. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev. 2014; 28: 87-94.