
Case Report
Ann Hematol Oncol. 2018; 5(5): 1210.
A Fatal EBV Related Hemophagocytic Lymphohistiocytosis (HLH) in a Young Patient
Magliano G¹*, Hohaus S¹, Cuccaro AR¹, D’Alo’ F¹, Maiolo E¹, Giustiniani MC² and Bacigalupo A¹
1¹Department of Hematology, Fondazione Policlinico Universitario Gemelli IRCCS, Universita’ Cattolica del Sacro Cuore, Rome, Italy
²Department of Pathology, Fondazione Policlinico Universitario Gemelli IRCCS, Universita’ Cattolica del Sacro Cuore, Rome, Italy
*Corresponding author: Magliano G, Department of Hematology, Fondazione Universitaria Policlinico Gemelli-IRCCS, Universita’ Cattolica del Sacro Cuore, Italy
Received: June 18, 2018; Accepted: July 30, 2018; Published: August 06, 2018
Abstract
We describe the case of a 25-year old African male patient, who developed a fatal EBV-related hemophagocytic lymphohistiocytosis (HLH). There was no evidence of previous infectious mononucleosis. HLH was diagnosed according to current criteria.
There was no response to treatment with rituximab, etoposide and cyclosporine with progressive worsening of the clinical conditions, early multiorgan failure, with >1 × 106 EBV copies/ml, and death. Autopsy revealed multi-organ localization of the disease in the liver, spleen and marrow, further confirming the refractoriness of this catastrophic EBV-related HLH, in a young immunocompetent patient.
Keywords: Epstein barr virus; Hemophagocytic lymphohistiocytosis (HLH); Cyclosporine; Pancytopenia
Case Presentation
We present the case of a 25-year-old African patient who developed a fatal EBV-related hemophagocytic lymphohistiocytosis and who was admitted, on arrival in Italy from Mali, in November 2016. His medical history revealed malaria infection in childhood. The patient was pancytopenic, with elevated ferritin, LDH, triglycerids and bilirubin.
Specific tests for Plasmodium malariae, Leishmania, Schistosoma, Borrelia, Leptospira and Rickettsiae were negative.
On admission to our Hematology Department the patient was febrile, with scleral jaundice, splenomegaly (21 cm) and a right pleural effusion.
Laboratory evaluation confirmed a severe pancytopenia: haemoglobin 6.1 g/dL, platelets 17x109/L and leukocytes0.17x109/L. LDH was 1783 IU/L, total bilirubin was 2.4 mg/dL and triglicerids were 335 mg/dL (Table 1).
![]()
On Admission
At the time of death Normal range
Haemoglobin
6.1 g/dL
6.7 g/dL
12-14 g/dl
Platelet count
17x109/L
6x109/L
150-450 X 109/l0,28x109/L
WBC
0.17x109/L
4-10 X 109/l
Chemistry
LDH
1783 UI/L
2949 UI/L
230-460 UI/l
GPT
45 UI/L
313 UI/
7-45 UI/l
Creatinine
0.62 mg/dL
0.86 mg/dL
0.7-1.2 mg/dl
Total bilirubin
2.4 mg/dL
10 mg/dL
0.3-1.2 mg/dl
Tryglicerides 335 mg/dL
270
20-170 mg/dL
Ferritin
16500 ng/mL
na
11-307 ng/ml
Haptoglobin
7.75 mg/dL
na
45-320 mg/dL
Albumin
25 g/L
22 g/L
34-48 g/L
Total proteins
56 g/L
46 g/L
65-85 g/L
Coagulation tests
aPTT
38.3 sec
73.2 sec
20-38 sec
Fibrinogen
193 mg/dL
180 mg/dL
200-400 mg/dL
INR
1.31 sec
2.11 sec
0.8-1.2 sec
D-Dimerus
2453 ng/mL
4865 ng/mL
<500 ng/mL
Abbreviations: WBC: White Blood Cells; LDH: Lactate Dehydrogenase; GPT: Glutamic Pyruvic Transaminase; aPTT: Activated Partial Thromboplastin Time; INR: International Normalized Ratio; na: Not Available
Table 1: Laboratory findings during hospitalization.
In addition there was a high PCR (19.8 mg/L), hypoproteinemia and hypoalbuminemia and ferritin was 16.500 ng/ml. On the second day of hospitalization IgG anti CMV and IgG anti-EBNA antibodies were together positive with 336.916 EBV copies/mL (Figure 1).
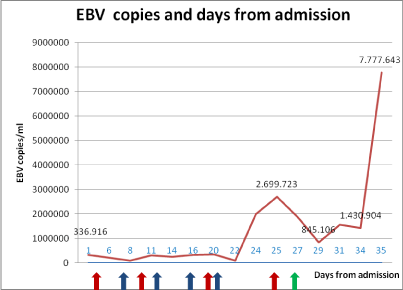
Figure 1: EBV copies and days from admission.
Red arrows: Rituximab; Blue arrows: Etoposide; Green arrows: Cyclosporine
Bone marrow histology showed high cellularity (activated macrophages and lymphoplasmacellular elements) with erithro and hemophagocytosis.
On the basis of the criteria of the Histiocyte Society, we diagnosed an acquired EBV related HLH and started chemotherapy with four cycles of rituximab (375 mg/m2) and four cycles of etoposide (150 mg/m2).
In spite of chemotherapy, the chest x-ray showed a right parailarhypodiaphany and a omolateralpleuric effusion.
The patient spiked daily temperature >39 °C, and remained pancytopenic, requiring transfusion support.
After 20 days of treatment elevated transaminases were detected with decreasing serum albumin. We faced increasing of inferior limbs edema, associated with edema of the right upper limb, as progression of an anasarcatic status. On day 25 EBV genome rised to 2.699.723 copies/mL. A fourth cycle of rituximab was then performed.
Despite the high level of EBV, cyclosporine (150 mg x2/die) intravenously was then started on day 27.
On day 34 of hospitalization the patient went to cardiorespiratory arrest. Cardiopulmonary resuscitation was performed. After reanimation cerebral CT revealed a focal hypodensity in the right-parapontine region.
The clinical conditions rapidly worsened and the patient was transferred to the ICU. EBV load had increased to 7.777.643 copie/ mL. The patient died 35 days after admission.
Autopsy revealed aspects related to hemophagocityc syndrome in multiple organs (liver, spleen and bone marrow) (Figure 2&3). The spleen was 23x15cm, and necrotic areas were found in the subcapsular area. Furthermore, wide bilateral serouspleuric effusion and massive alveolar-interstitial hemorrhage were found in the lungs.
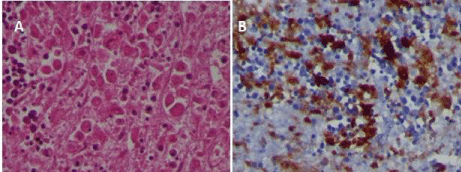
Figure 2: Overall survival, autologous stem cell transplant (ASCT) versus no ASCT (p=0.12).
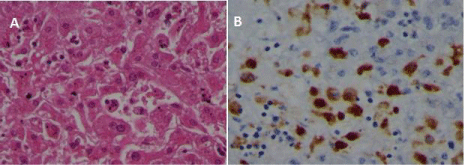
Figure 3: Liver autopsy. Numerous macrophages, some showing hemophagocytosis, are present in sinusoids (A) Hematoxylin-eosin, stained with antibody for
CD68 (B).
Discussion/Conclusion
Hemophagocytic lymphohistiocytosis (HLH) is a rare lifethreatening clinical syndrome of immune dysregulation with tissue infiltration of cytokine-activated macrophages (histiocytes), as well as cytotoxic T-lymphocytes. Secondary-or acquired- HLH (SHLH) is commonly observed in adults, associated with several underlying triggering conditions, such as autoimmune disorders, malignancies, and mostly to various forms of infection [1,2]. SHLH is reported to be fatal in 22-59% of patients [3] in spite of specific therapies. SHLH is triggered by infections in 50% of reported cases, viral infections being the most frequent cause, either as a primary infection in healthy people or reactivated infections in immunosuppressed patients [4,5]. Epstein-Barr virus (EBV) has been recognized as the leading infectious cause of HLH [1]. However, in infectious mononucleosis the virus infects B cells, whereas in EBV-associated HLH the virus genome has been detected both in T and B cells [6]. In EBV-associated HLH, a high viral DNA load has been associated with poor outcome [7]. Early diagnosis of HLH is crucial to institute therapy and for this reason the Histiocyte Society revised the diagnostic criteria in 2004 [8] (Table 2).
![]()
1) Molecular diagnosis of familial haemophagocytosis (Pathologic mutations of Perforin (PRF1), SH2D1A/SAP, UNC13D, Syntaxin 11 (STX11), MUNC18-2, Ras-related Protein Rab 27a (RAB27a)
Or
2) Five out of eight diagnostic criteria listed below are fulfilled:
1. Fever ≥38.5°C
2. Splenomegaly
3. Cytopenias (affecting at least 2 of 3 lineages in the peripheral blood):
Hemoglobin <9 g/dL (in infants <4 weeks; hemoglobin <10 g/dL)
Platelets <100.000/mL
Neutrophils <1000/mL
4. Hypertrigliceridemia(fasting >265 mg/dL) and or hypofibrinogenemia (>150 mg/dL)
5. Hemophagocytosis in bone marrow or spleen or lymph nodes or liver
6. Low or absent NK.cell activity
7. Ferritin >500 ng/ml
8. Elevated soluble CD25 (alpha chain of soluble IL-2 receptor)
Table 2: Diagnostic criteria for hemophagocytic syndrome as proposed by the Histiocyte Society: one of two criteria should be present for diagnosis [8].
We are here presenting a case of fatal EBV-associated HLH in a young patient, and would like to discuss early diagnosis, treatment and outcome. As to the first point, on admission to another Centre, the diagnostic work-up aimed to exclude infections which are endemic in Mali, such as malaria, known to be the only previous disease of the patient. The patient had five out of eight HLH- 2004 criteria (splenomegaly, pancytopenia, hypertriglyceridemia, hyperferritinemia, fever) even without bone marrow biopsy. Indeed, in half of HLH patients, the initial bone marrow biopsy may only show erythroid hyperplasia, leading to wrongly reject the diagnosis [9].
Simple laboratory tests may be helpful for an early diagnosis of secondary HLH and would prevent a diagnostic delay, even in smaller hospitals. Using the scoring system by Fardet [9], our patient had a score of 210 points and a 93% probability of having HLH, on first admission.
There were signs of early organ damage (pleural effusion, hepatosplenomegaly): similarly, Berry and coworkers reported two 18-years old patients, who were first admitted with acute liver failure, DIC, pancytopenia and cardio-respiratory arrest due to EBV related HLH. HLH should be suspected when young adults develop acute bone marrow failure and multiple organ dysfunctions [10].
As to treatment, we were facing an EBV infection and we thus started with rituximab, and followed with the HLH 2004 protocol, with four courses of dexamethasone and etoposide. Rituximabcontaining regimens have proved to significantly reduce EBV load and signs of inflammation [11]. Etoposide was given regardless of blood counts, as early administration has been related to a better outcome [3]. Etoposide induces apoptosis of activated immune cells and limits the release of cytokines, but also inhibits EBV nuclear antigen synthesis and transformation of EBV-infected cells [12]. Despite combined treatment the clinical conditions deteriorated, and the EBV load increased. The HLH- 2004 protocol calls for cyclosporine to start early during induction therapy [8]. The reason to delay cyclosporine in our case was the extremely high level of EBV DNA. When we did start cyclosporine, much in despair, we did see a significant increase in WBC from 0.64x109/L to 2.2x109/l. To our knowledge, management of cyclosporine in aggressive EBV -related HLH has not been clearly defined.
As to the catastrophic outcome, on the day of death the hemoglobin level was 6.7 g/dL, platelet counts 6x109/L, and WBC 0.28x109/L, total bilirubin was 10 mg/dL and GPT was 685 UI/L. The EBV load increased seven fold in the last 24 hours of life, up to 7.777.643 copies/mL.
The aggressive HLH was proven by multiple localization of hemophagocytosis. In particular, significant HLH was shown in the liver, which both in the parenchyma and in the sinusoids.
At present, there are little data regarding potential salvage therapies for patients who do not respond to induction therapy. Notably, Marsh and coworkers treated 22 pediatric and adult refractory HLH with subcutaneous alemtuzumab: 77% survived and were allografted [13].
We have treated this young patient two months after the onset of first symptoms: we hypothesize that the outcome would have been different with an early recognition and treatment. We believe that both the EBV infection and the related HLH were firmly established at the time the patient started effective therapy, and this may have been a strong predictor of the negative outcome.
EBV related HLH can be fatal even in the young immunocompetent patient: the potential aggressiveness of the disease suggests that early diagnosis and early therapy are mandatory. Possible salvage therapies are needed for refractory HLH, as a bridge to transplant, and this may improve the dismal prognosis.
References
- Hashemi-Sadraei N, Vejpongsa P, Baljevic M, Chen L, Idowu M. Epstein- Barr Virus-Related HemophagocyticLymphohistiocytosis: Hematologic Emergency in the Critical Care Setting. Case Rep Hematol. 2015.
- Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with hemophagocytic syndrome. Lancet Infect Dis. 2007; 7: 814-822.
- Arca M, Fardet L, Galicier L, Rivière S, Marzac C, Aumont C, et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015; 168: 63-68.
- Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014; 383: 1503-1516.
- Chen J, Wang X, He P, Li Y, Si M, Fan Z, et al. Viral etiology, clinical, and laboratory features of adult hemophagocyticlymphohistiocytosis. J Med Virol. 2016; 88: 541-549.
- Kasahara Y, Yachie A, Takei K, Kanegane C, Okada K, Ohta K, et al. Differential cellular targets of Epstein-Barr virus (EBV) infection between acute EBV-associated hemophagocyticlymphohistiocytosis and chronic active EBV infection. Blood. 2001; 98: 1882-1888.
- Kimura H, Hoshino Y, Hara S, Nishikawa K, Sako M, Hirayama M, et al. Viral Load in Epstein-Barr Virus-Associated Hemophagocytic Syndrome.Microbiol Immunol. 2002; 46: 579-582.
- Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocyticlymphohistiocytosis. Pediatr Blood Cancer. 2007; 48: 124- 131.
- Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014; 66: 2613- 2620.
- Berry PA, Bernal W, Pagliuca A, Sizer E, Salisbury JR, Wendon JA, et al. Multiple organ failure and severe bone marrow dysfunction in two 18 year-old Caucasian patients: Epstein–Barr virus and the haemophagocytic syndrome. Anaesthesia. 2008; 63: 1249-1254.
- Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus induced haemophagocyticlymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013; 162: 376-382.
- Schram AM, Berliner N. How I treat hemophagocyticlymphohistiocytosis in the adult patient. Blood. 2015; 125: 2908-2914.
- Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocyticlymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013; 60: 101-109.