
Case Report
Ann Hematol Oncol. 2019; 6(3): 1239.
AML in Mayer-Rokitansky-Küster-Hauser Syndrome
Al-Rabi K¹*, Obeidat A¹ and Alsmadi O²
¹Department of Medical Oncology, King Hussein Cancer Center, Jordan
²Department of Cell Therapy and Applied Genomics, King Hussein Cancer Center, Jordan
*Corresponding author: Kamal Al-Rabi, Department of Medical Oncology, King Hussein Cancer Center, Amman, Jordan
Received: December 28, 2018; Accepted: February 12, 2019;Published: February 19, 2019
Abstract
The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is the second most common cause of primary amenorrhea, it is characterized by congenital hypoplasia of the uterus and the upper part of the vagina. The incidence of MRKH syndrome has been estimated as 1 in 4500 women [1]. Most of the studies suggest that MRKH syndrome has been considered as a genetic disease, and genes such as the HOXA7, HOXA9-13, HOXD9-13, and WNT4 have been considered as possible offenders [2].
Malignancies of ovaries, uterus and renal were reported in association with this syndrome [3-5]. Here we are reporting the first case of MRKHS associated with AML in literature.
Keywords: Mayer-Rokitansky-Küster-Hauser syndrome; AML
Introduction
Mayer-Rokitansky-Kuster-Hauser syndrome is characterized by congenital aplasia of the uterus and the upper two thirds of the vagina in women showing normal developmental of secondary sexual characteristics, normal ovarian function and normal 46, XX karyotype [1]. Type one is the isolated form [6]. The second one (type II) is an atypical syndrome, with the presence of asymmetric uterine remnants and abnormal uterine tubes. Such syndrome type may be associated with ovarian disease, congenital renal, bone abnormalities and hearing defects. A third one, the so called MURCS type, involves uterovaginal hypoplasia or aplasia, renal, bone and cardiac malformations [1,7]. The extent of vaginal aplasia varies, ranging from virtually absent to virtually inconsequential. MRKH syndrome usually remains undetected until the patient presents with primary amenorrhea despite normal female sexual development. MRKH syndrome is the second most common cause of primary amenorrhea [1].
Cases of ovarian, uterine and renal cell carcinoma were reported in association with MRKH [3-5]. However, MRKH has never been previously described to be associated with AML. Here we are presenting a case of 21 year old Caucasian female with MRKH syndrome found to have AML- M2 with t (6;9).
Case Report
A 21-year-old single female presented to King Hussein cancer center after being diagnosed with Acute Myeloid Leukemia outside the center, she was accidently found to be anemic during routine blood work to investigate sparse scalp hair, otherwise she was asymptomatic. Complete blood count showed thrombocytopenia, platelets of 89,000/μl and anemia Hgb of 8.5g/dl, WBC 11.9×10 /μl,. Blood film revealed numerous blasts in peripheral blood with giant platelets, kidney function, liver function and coagulation tests were all within normal range. In the light of these results bone marrow biopsy was done and showedhypercellular for age. The estimate M:E ratio is 8:1. Infiltration of bone marrow with 26% blasts (Figure 1). Flowcytmetry showed myeloblasts which expresses CD34, CD 117, CD33, CD13, CD38, CD58, HLA-DR and cMPO.
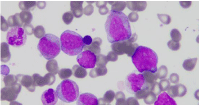
Figure 1: Wright-Giemsa stain of bone marrow aspirate (100 X) showing
at least three myeloblasts. These blasts are large in size, have nuclei with
open chromatin, multiple nucleoli, and moderate amounts of finely granular
cytoplasm. The remaining myeloid cells show different stages of maturation.
Myeloblasts in this marrow represented 26% of cells.
Upon evaluation and counseling for fertility preservation, she gave a history of primary amenorrhea, which was first investigated at age of 16 years and was diagnosed with MRKHS Type 1. Pelvic ultrasound showed that the uterus is hardly seen and appears hypoplastic, both ovaries were prominent in size with dominant follicle in the right ovary, pelvic MRI showed there was marked uterine hypoplasia with hypoplastic vagina, the endometrium was not visualized the urinary bladder was normal, both ovaries appear unremarkable, there was marked uterine hypoplasia with hypoplastic vagina, the endometrium was not visualized (Figure 1). Reproductive hormones levels FSH: 3.43mIU/mL (0.3 to 10.0 IU/L), LH: 7.2IU/L follicular phase of the menstrual cycle: 1.9 to 12.5 IU/L. women at the peak of the menstrual cycle: 8.7 to 76.3 IU/L. women in the luteal phase of the menstrual cycle: 0.5 to 16.9 IU/L and total testosterone was 0.241 15 to 70 ng/dL.
Regarding her family history, her parents are first cousins to each other. She has 4 aunts, three of them are infertile; one was recently diagnosed with endometrial cancer and the other was diagnosed with endometriosis. None of them has primary or secondary amenorrhea as well as MRKHS. Her father’s aunt had primary amenorrhea but passed away without definitive diagnosis, she had no family history of acute leukemia or hematological disorders.
Cytogenetics examination 46, XX, t (6;9) (p22;q34) (Figure 3), Fluorescent in Situ Hybridization (FISH) examination was negative for t (8;21), FLT3 mutation by Polymerase Chain Reaction (PCR)was negative.
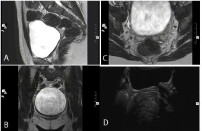
Figure 2: (a). T2WI Sagital Pelvic MRI showing the hypoplastic uterus and
Vagina. (b) Axial High Resolution T2WI showing the hypoplastic uterus. (c)
High Resolution Coronal T2WI showing both ovaries. (d) Pelvic Ultrasound
showing the hypoplastic uterus.
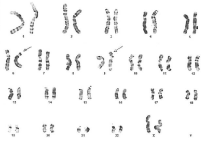
Figure 3: Representative karyotype demonstrating the translocation between
6p22 and 9q34. Arrows point to the derivative chromosomes 6 and 9.
She received 7+3 induction (standard-dose cytarabine, and 3 days of an anthracycline) on 9/11/2017, which was followed by uneventful course and she achieved complete remission morphologically on 29/11/2018. then she was consolidated with 2 cycles of High dose Ara-C, Human Leukocyte Antigen (HLA) typing showed no fully matched related donor, so she underwent Haploidentical Allogenic Stem cell Transplant from her sister on 12/03/2018, bone marrow biopsy on D+30 after transplant was in complete remission, chimerism was 94%, her post-transplant course was uneventful except for CMV reactivation and was treated with Ganciclovir, she is still under follow up by transplant team.
Discussion
Acute Myeloid Leukemia (AML) is characterized by the clonal proliferation of hematopoietic stem or progenitor cells and results in a failure of normal hematopoiesis [PubMed: 20008237]. AML properties such as self-renewal, apoptosis-resistance, and failure of cell differentiation, is mediated in some cases by aberrant fusion proteins [PML-RARa, AML1–ETO, inv(16), t(1:14)] and, in others by proliferation signals driven by mutated genes, e.g. FLT3-ITD and NPM1 [9]. Wingless-Type (WNT) pathway on the other hand is implicated as key component of hematopoietic events during early embryogenesis [10], with role in determining cell fate, proliferation, migration, polarity, and gene expression [11]. WNT family genes appear to signal mainly through the canonical Wnt/β-catenin pathway, including the WNT-4 gene [12]. The dysregulation of WNT canonical pathway is tightly linked to expression of AML patients [13]. WNT4 expression was also shown to be strongly reduced in leukemic cell lines, and in blast cells derived from patients with leukemia, relative to the expression in healthy individuals’ mature blood cells [14]. Disturbance of WNT signaling is linked to many human disorders, including birth defects and cancers. Epigenetic regulation of the WNT pathway was predictive to relapse in a subgroup of AML patients too [15]. From the above, it is clear WNT signaling is central to AML pathogenesis.
Mayer Rokitansky Kuster Hauser (MRKH) syndrome, also known as Müllerian agenesis, is the second most common cause of primary amenorrhea, characterized by congenital absence of major female sex organs including the uterus, cervix, and the upper part of the vagina in otherwise kayotipically normal females (46, XX) [16]. MRKH syndrome incidence is about 1/4, 500- newborn females [1]. It is clinically heterogeneous and can be divided into two subtypes: MRKH type 1, in which only the upper vagina, cervix and the uterus are affected; MRKH type 2 however is characterized by additional abnormalities affecting both renal and skeletal systems [7]. MRKH syndrome is mostly sporadic, but can be heritable in some cases, with an autosomal dominant inheritance pattern, with incomplete penetrance and variable expressivity [17]. Genes such as the HOXA7, HOXA9-13, HOXD9-13 and WNT4 have role in embryonic origin of the female reproductive tract [18-20]. Genomic rearrangements had been also identified in MRKH-affected patients, which was also confirmed in mouse animal models [21]. Nevertheless, WNT4 has been the most associated gene with MRKH in several reports on human subjects. WNT4 is critical for the development of the female reproductive tract. Mutated WNT4 was reported to be associated with Müllerian-Duct regression and virilization in an otherwise cytogenetically normal woman (46, XX) [22]. Mutations in WNT4 leading to Loss-of-function in WNT4 have been identified in 46, XX females as well, with Mullerian duct abnormalities and androgen excess [23].
The patient we reporting here, presented clinically with a classical type I MRKH. Considering she is a daughter to a consanguineous marriage, with multigenerational amenorrheic relative females, we like to speculate her condition is a heritable one. In addition, the unusual combination of this MRKH syndrome with AML is very surprising and deserves further critical clinical and molecular characterization. While it would be possible to tackle a small set of candidate genes including the ones cited above, it would be perhaps more inclusive to conduct whole exome sequencing on this patients with hope the genetic defect(s) underlying these two independent clinical presentations is/are discovered, which we shall pursue as follow-up on this patient.
Conclusion
This the first case in literature to report the occurrence Acute myeloid leukemia in Mayer RokitanskyKuster Hauser (MRKH) syndrome, WNT4 has been the most associated gene with MRKH ,Wingless-Type (WNT) pathway on the other hand is implicated as key component of hematopoietic events during early embryogenesis, in addition it is dysregulated in AML cells.
Consent: Consent has been taken from the patient.
Disclaimer: The abstract has not been previously presented or published in any conference. The manuscript was not part of any research, PhD or thesis project.
References
- Morcel K, Guerrier D, Watrin T, Pellerin I, Levêque J. “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: clinical description and genetics,” Journal de Gynécologie Obstétrique et Biologie de la Reproduction. 2008; 37: 539-546.
- Sultan C, Biason-Lauber A, Philibert P. “Mayer-Rokitansky-Kuster-Hauser syndrome: Recent clinical and genetic findings”. Gynecological Endocrinology. 2009; 25: 8-11.
- Mermerkaya M, Burgu B, Hamidi N, et al. Mayer-Rokitansky-Küster-Hauser syndrome accompanied by renal cell carcinoma: a case report. J Pediatr Hematol Oncol. 2013; 35: e309-e310.
- Ghirardini G, Magnani A. Mayer-Rokitansky-Küster-Hauser syndrome and ovarian cancer. Report of a case. Clin Exp Obstet Gynecol. 1995; 22: 247-248.
- Fukuda J, Kumazawa Y, Figimoto T, Tanaka T. Mayer-Rokitansky-Kuster-Hauser syndrome complicated by either uterine leiomyoma or ovarian tumor. J Obstet Gynaecol Res. 2010; 36: 191-194.
- Jones KL. Rokitansky sequence. Smith's recognizable patterns of human malformations (Fourth ed). Edited by: Saunders WB. Philadelphia. 1988; 570-571.
- Strübbe EH, Willemsen WN, Lemmens JA, Thijn CJ, Rolland R. Mayer-Rokitansky-Küster-Hauser syndrome: distinction between two forms based on excretory urographic, sonographic, and laparoscopic finding. AJR Am J Roentgenol. 1993; 160: 331-334.
- Steffen B, Müller-Tidow C, Schwäble J, Berdel WE, Serve H. The molecular pathogenesis of acute myeloid leukemia. Crit Rev Oncol Hematol. 2005; 56: 195-221.
- Kielman MF, Rindapää M, Gaspar C, van Poppel N, Breukel C, van Leeuwen S, et al. Apc modulates embryonic stem-cell differentiation by controlling the dosage of beta-catenin signaling. Nat Genet. 2002; 32: 594-605.
- Khan NI, Bendall LJ. Role of WNT signaling in normal and malignant hematopoiesis. Histol Histopathol. 2006; 21: 761-774.
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009; 17: 9-26.
- LeMajeti R, Becker MW, Tian Qe TL, Yan X, Liu R, Chiang JH, et al. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc Natl Acad Sci U S A. 2009; 106: 3396-3401.
- García-Castro B, Alvarez-Zavala M, Riveros-Magaña AR, Ortíz-Lazareno PC, Ratkovich-González S, Hernández-Flores G, et al. Restoration of WNT4 inhibits cell growth in leukemia-derived cell lines. BMC Cancer. 2013; 13: 557.
- Valencia A, Román-Gómez J, Cervera J, Such E, Barragán E, Bolufer P, et al. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia. 2009; 23: 1658-1666.
- Morcel K, Camborieux L; Programme de Recherches sur les Aplasies Müllériennes, Guerrier D. Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2007; 2:13.
- Fontana L, Gentilin B, Fedele L, Gervasini C, Miozzo M. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin Genet. 2017; 91: 233-246.
- Mendelsohn C, Lohnes D, Décimo D, Lufkin T, LeMeur M, Chambon P, et al. Function of the Retinoic Acid Receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994; 120: 2749-2771.
- Kastner P, Mark M, Ghyselinck N, Krezel W, Dupé V, Grondona JM, Chambon P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development. 1997; 124: 313-326.
- Burel A, Mouchel T, Odent S, Tiker F, Knebelmann B, Pellerin I, et al. Role of HOXA7 to HOXA13 and PBX1 genes in various forms of MRKH syndrome (congenital absence of uterus and vagina). J Negat Results Biomed. 2006; 5: 4.
- Fontana L, Gentilin B, Fedele L, Gervasini C, Miozzo M. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin Genet. 2017; 91: 233-246.
- Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ. A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46, XX woman. N Engl J Med. 2004; 351: 792-798.
- Philibert P, Biason-Lauber A, Rouzier R, Pienkowski C, Paris F, Konrad D, et al. Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and müllerian duct abnormalities: a French collaborative study. J Clin Endocrinol Metab. 2008; 93: 895-900.