
Review Article
Ann Hematol Oncol. 2019; 6(4): 1245.
JAK/STAT Signaling Pathway and Inhibitors in Inflammatory and Neoplasmatic Skin Diseases
Karagianni F1, Kolialexi A2, Papadavid E1*
12ndDepartment of Dermatology-Venereology, ATTIKON University Hospital, National and Kapodistrian University of Athens Medical School, Greece
23rdDeparmtent of Obstetrics and Gynecology, National and Kapodistrian University of Athens Medical School, Greece
*Corresponding author: Papadavid Evangelia, 2nd Department of Dermatology-Venereology, ATTIKON University Hospital, National and Kapodistrian University of Athens Medical School, Tel +30 210 5832368, 5832396, Fax +30 210 5832397, Greece
Received: February 04, 2019; Accepted: March 11, 2019;Published: March 18, 2019
Abstract
Novel therapeutic targets have been unraveled during the past years and one major target is the JAK/STAT pathway, which is involved in processes such as immunity, cell division, cell death and tumour formation. Since the role of the JAK/STAT pathway has been investigated in several aspects of human diseases, it is of great importance to understand the molecular mechanisms of JAK/STAT that implicated in inflammatory and neoplasmatic skin diseases and to target this signaling pathway for therapeutic purposes. In this review, the scientific literature has been evaluated in order to well define the mechanisms behind the inflammatory (atopic dermatitis, psoriasis) and neoplasmatic skin disease (cutaneous T-cell lymphoma), the role of the microenvironment and the current use of specific inhibitors in order to block the signaling of this particular pathway targeting better treatment in the future either with the use of new molecules or already known with different drug combinations.
Keywords: Jak/Stat pathway; Inhibitors; Atopic dermatitis; Psoriasis; Cutaneous T-cell lymphoma
Introduction
JAK/STAT pathway
Recent years have brought great progress in our understanding of the pathogenesis of inflammatory and neoplasmatic diseases uncovering novel therapeutic targets. One of these newly identified targets is the Janus Kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) pathway, which is fundamental for the downstream signaling of inflammatory cytokines and of different growth factors.
The JAK/STAT pathway is an ancient pathway, evolutionary conserved, which has yielded fundamental insights about cellular communication and the role of membrane to nucleus signaling in controlling gene expression. It has also shaped our understanding of the mammalian immune system. It is a signaling cascade crucial for embryonic development, cell growth, haemopoietic development and differentiation, innate and adaptive immunity and the inflammatory response [1]. The JAK family consists of four Jaks, (JAK1, JAK2, JAK3, and TYK2), which selectively bind different receptor chains that lack intrinsic enzymatic activity [2,3]. The cytokine receptors are shown in Table 1 and the most important is the type I cytokines family receptors. They have four a-helical bundle structures and include many interleukins, as well as some growth and haematopoietic factors. One important family of type I cytokines is the common cytokine-receptor γ-chain (γc) family, which consists of interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21, and is so named because the receptors for these cytokines share γc (Figure 1). IL-2 functions as a T cell growth factor, can augment NK cell cytolytic activity, and promotes immunoglobulin production by B cells [4]. In addition, it contributes to the development of regulatory T (Treg) cells and therefore peripheral T cell tolerance [5] as well as regulating the expansion and apoptosis of activated T cells [6]. IL-4 is required for the development and function of T helper 2 (Th2) cells and is therefore regarded as the classical Th2-type cytokine. IL-4 also has an important role in allergy and immunoglobulin class switching [7]. IL-7 has a central role in the development of T cells in both humans and mice. IL-9 is produced by a subset of activated CD4+ T cells [8,9] and induces the activation of epithelial cells, B cells, eosinophils, and mast cells [10]. IL-15 also has an essential role in CD8+ T cell homeostasis [11]. IL-21 is the most recently described member of this family [12], and it has broad actions that include promoting terminal B-cell differentiation to plasma cells, cooperating with IL-7 or IL-15 to drive the expansion of CD8+ T cell populations, and acting as a pro-apoptotic factor for NK cells and incompletely activated B cells [12].
![]()
Cytokine/Cytokine receptor families
Example
Associated JAKs
Type I interferons
IFN-a
JAK1, TYK2
Type II interferons
IFN-γ
JAK1, JAK2
Il12 receptor β1 cytokines
IL23
JAK2, TYK2
gp130 receptor cytokine family
IL6
JAK1, JAK2, TYK2
IL10 family cytokines
IL22
JAK1, JAK2, TYK2
Common γ chain cytokines
IL4
JAK1, JAK3
βc receptor cytokine family
GM-CSF
JAK2, TYK2
Homodimeric receptor cytokine family
EPO
JAK2
Table 1: Cytokines and associated JAKs.
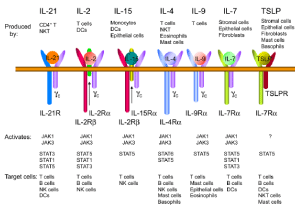
Figure 1: Receptors for γc family cytokines. Receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21, expressed by a variety of immune cells, activate JAK1 and JAK3
and target at different immune cells. (Adapted from Yrina Rochman, Rosanne Spolski, and Warren J. Leonard, 2009).
Ligand binding induces dimerization of the receptor/kinase complexes, thus activating the kinases to phosphorylate each other as well as the receptor. Therefore, they bind cytosolic domains of these receptors and are activated by cytokine receptor engagement by cognate ligand. The active JAKs phosphorylate each other as well as the intracellular tail of the receptor subunits, creating docking sites that recruit downstream signaling molecules. Jak-mediated phosphorylation activates STATs, which in turn directly bind DNA and regulate specific gene expression [13].
In mammals, JAK1, JAK2, and TYK2 are expressed ubiquitously whereas JAK3 is primarily expressed in hematopoietic and lymphoid tissues cells as well as in vascular muscle cells [1,13-19].
In normal cells JAK/STAT signaling is transient and tightly regulated by multiple negative regulatory mechanisms, including activity of the SOCS (suppressors of cytokine signaling) family of endogenous JAK inhibitors. In addition, SOCS proteins facilitate proteosomal degradation of activated JAKs. Furthermore, activation of the JAK/STAT pathway is controlled by phosphatases, which dephosphorylate and inactivate JAKs and STATs including SHP- 1, and proteins belonging to the PIAS family that block binding of STATs to DNA. It is generally accepted that TYK2, JAK2, and STAT4, which are IL-12 signaling pathway components, are essential for Th1 cell differentiation, while JAK1, JAK3, and STAT6, which are IL-4 signaling components, are critical for Th2 differentiation. In addition, STAT5A/B, which are involved in the upregulation of GATA3 and IL-4Ra, and STAT3, which helps STAT6 bind to its target genes, also play some roles in Th2 cell differentiation [19-21]. Effective innate and adaptive immune responses require functional Jak signaling to protect the organism from infections or tumors and mutations leading to loss of function make up some of the commonest inherited severe immunodeficiencies. Conversely, activating mutations or mutations leading to functional loss of Jak members cause malignant transformation of lymphocytes or myeloid cells. Point mutations either predicted or confirmed to be gain-of-function are reported in JAK1 (0.9% of cases), JAK3 (2.7%), STAT3 (0.9%), and STAT5B (3.6%) [22]. Hyperactive JAK and STAT proteins often result from structural changes in their functional domains. For instance, amino acid substitutions in the pseudokinase domain of JAK proteins enhance their tyrosine kinase activity, while changes in the Src homology 2 (SH2) domain of STAT proteins are predicted to enhance dimerization. In addition, copy number gains of JAK2 (13% of cases), STAT3 (60%), and STAT5B (60%) are frequent and have been shown to correlate with increased expression [22]. In NGS studies, it was demonstrated that CTCL cell lines bearing gain-of-function point mutations in JAK3 are sensitive to these compounds, opening the possibility for their potential use in CTCL patients carrying JAK mutations [23,24].
Deregulation of cytokines is known to contribute to diverse inflammatory and immune-mediated disorders. Deregulation of JAK/STAT signaling has been implicated in inflammatory diseases such as rheumatoid arthritis, atopic dermatitis, psoriasis as well as in a variety heamatological malignancied (myeloid disorders) and neoplasmatic diseases of skin such as cutaneous T cell lymphoma.
Numerous cytokines rely on the JAK-STAT pathway for signaling, and new and effective small molecule inhibitors have been developed for a range of disorders [25]. Selective JAK-inhibitors influence the phosphorylation and activation of different JAKs, which results in the blockage of the cascade of inflammatory cytokines [26].
In this review, we underline the importance of JAK/STAT pathway in inflammatory and neoplastic skin diseases with an emphasis on the implicated mechanisms in the development of neoplasmatic skin disease and the current use of specific inhibitors in order to block the signaling of this particular pathway.
The selective usage of JAKs by different receptors explains their distinct roles and becomes particularly important with the generation of pharmacological inhibitors when specific or relatively discrete functional outcomes are sought (Table 1). The realization that JAKs contribute substantially to the immunologic processes in autoimmune and neoplastic diseases led to the development of JAK inhibitors as therapeutic immunosuppressive agents [27,28].
JAK/STAT pathway in skin diseases
Inflammatory diseases
Atopic Dermatitis (AD): AD is a common inflammatory skin disease characterized by chronic inflammation and skin infiltration of inflammatory cells including predominantly lymphocytes, eosinophils, and mast cells. This multifactorial disease possible results from a complex crosstalk between genetic and environmental factors. The factors that lead to AD development are the barrier dysfunction and abnormal immune activation of T helper Th2, Th22, and varying degrees of Th1 and Th17 among various subtypes.
Filaggrin gene mutation defects is a cause of barrier dysfunction in some AD patients, but IL-4 is also able to downregulate barrier proteins filaggrin, loricrin and in volucrin through the JAK-STAT pathway making the epidermis more penetrable by allergens and pathogens [27]. Once penetrated through the epidermis, allergens/ pathogens are detected by dendritic cells, which become subsequently activated to present these antigens to naïve Th0 cells. The naïve Th0 cell can then differentiate into the Th2 cell through the JAK1, 3-STAT6 pathway under the influence of IL-4. In the Th0 cells, the STAT6 pathway can also upregulate GATA3, a master regulator of Th2 cells. GATA3 in turn suppresses Foxp3, the master regulator in Treg cells, thus allowing more T cells to be activated [27].
Exaggerated Th2 response, disruption of the epidermis barrier functions, high levels of serum IgE, and decreased production of Antimicrobial Peptides (AMPs) are the key findings in AD [28,29]. Since AD is a Th2-dominant disease, examination of how the JAK-STAT pathway regulates Th2 differentiation would help us to understand the possible roles that JAK-STAT play in AD. It is well established that IL-4 promotes the differentiation of Th2 cells, which sequentially produce IL-4, IL-5, IL-10, and IL-13. Th2 cells play a significant role by their abilities to provide IL-4 and IL-5 stimulation via the JAK-STAT pathway. The Th2 immune milieu is able to trigger epidermal cells to produce and release various chemokines (such as CCL26), pro-inflammatory cytokines, and angiogenic factors, leading to AD pathophysiology. In T cells, IL-4-activated STAT6 upregulates GATA3, the master regulator of Th2 cells. GATA3 in turn suppresses Foxp3, the master regulator in Treg cells [30,31]. This regulatory pathway could possibly explain why Treg function is suppressed in AD.
Inhibitors in AD: The importance of multiple type I/II cytokine receptor-using mediators in AD pathogenesis suggest that interfering with the JAK/STAT pathway could be a more potent therapeutic approach than neutralization of a single cytokine. Current studies have confirmed activation of JAK-STAT signaling within lesional skin of patients with atopy [32,33] and that inhibition of JAK JAK STAT signaling may ameliorate chronic dermatitides by improving skin barrier function [34,35]. Amano et al have shown in the NC/ Nga mouse model of AD, a topically applied pan-JAKi JTE-052 with nanomolar potency towards all JAKs to improve skin barrier function and induce filaggrin expression [35]. Currently, several JAKi are under investigation for the treatment of human AD. Up to now, there are seven JAK inhibitors used in clinical trials for AD in human patients either oral [upadacitinib (JAK1), PF-04965842 (JAK1), baricitinib (JAK1, JAK2), ASN002 (JAK/SYK), tofacitinib (JAK1, JAK3)] or topical [tofacitinib (JAK1, JAK3), ruxolitinib (JAK1, JAK2), delgocitinib (JAK1,2,3 and TIK2)] [36].
Psoriasis: Psoriasis is a chronic inflammatory disease of skin with genetic disposition that can be caused by either endogenous or exogenous triggering factors. The disease is characterized by mostly extend-sided, often symmetrically appearing erythematous plaques with white scaling [37]. In psoriasis, keratinocytes are regarded as the main targets of effector cytokines, such as IL-17 and IL-22, not a driver in the pathogenesis of this disease. External agents stimulate skin-resident DCs or other immune cells to induce their production of inflammatory cytokines, which activate keratinocytes [38-40]. These keratinocyte responses are considered to activate the development of the epithelial immune microenvironment in psoriatic inflammation and the subsequent propagation and chronicity of the disease at the interface of the body and the external environment.
Histologically, psoriasis is characterized by considerable thickening of the epidermis due to an increased proliferation of keratinocytes with a dense dermal infiltrate of immune cells, such as T Cells and Dendritic Cells (DCs). The pathophysiology of psoriasis is dominated by an IL-17A + Th17 immune response. Multiple cytokines that are highly expressed in psoriatic skin lesions signal via the JAK/STAT pathway, including IL-19, IL-20, IL-22 and IL-23 [41- 43]. Active STAT3 is typically present both in psoriatic immune cells and in psoriatic keratinocytes inducing epidermal hyperplasia [36- 45]. IL-23 is mainly produced by DCs and stimulation of the IL-23R in T cells leads to recruitment of JAK2/TYK2 and downstream to the activation of STAT3 [45,46] STAT3 is important for regulating the expression of IL-23R and for the expression of IL-17A, IL-17F and IL-22 in T cells [1].
Inhibitors in psoriasis: Tofacitinib is one Jak inhibitor that is implicated in most studies in psoriasis by inhibiting the expression of interleukin (IL)-23 and differentiation of T helper type 1 (Th1) cells. It is the most studied JAK inhibitor in moderate to severe plaque type psoriasis. Several phase III Randomized Controlled Trials (RCTs) have shown that significantly more patients achieve a 75% reduction in the Psoriasis Area and Severity Index (PASI 75) while on tofacitinib compared with placebo. These patients also demonstrated a dose-dependent improvement of PASI 75 on tofacitinib 10 mg twice daily as compared with 5 mg twice daily [46,47]. A phase III non-inferiority trial revealed that tofacitinib 10 mg twice daily was non-inferior to etanercept 50 mg twice weekly [48]. Another study of baricitinib demonstrated significantly more patients achieved PASI 75 as compared with placebo [49]. Topical JAK inhibitors for mild-moderate psoriasis have also been investigated. Tofacitinib showed variable results in a phase IIa trial. Differences in efficacy were speculated to be due to variability in moisturizing properties of the formulations tested [50]. Ruxolitinib (INCB018424) was studied in a non-blinded and nonvehicle- controlled trial and was found to reduce the mean area and severity of psoriatic lesions [51].
Neoplasmatic disease
Cutaneous T-cell Lymphoma (CTCL): Cutaneous T-cell lymphoma, a common subtype of primary T-cell lymphoproliferative disorder of skin, is a group of malignancies derived from skin-homing T cells representing a heterogeneous group of lymphoproliferative disorders with most derived from the CD4 helper/ inducer T cell subset [52,53]. The most common variants are the Mycosis Fungoides (MF) and Sezary Syndrome (SS).
MF is characterized by a usual long clinical course and mostly involvement of the skin. Early lesions of CTCL (MF) typically present as limited skin patches or plaques, showing various degrees of epidermotropism consisting of single or clusters (Pautrier microabscess) of atypical lymphocytes and a superficial band-like lymphoid infiltrate composed of small to medium-sized atypical T-cells with irregular, hyperconvoluted nuclei interspersed with small tumor-infiltrating T-cells and histiocytes [54]. A 25% of the early MF can progress to tumor stage [55].
At the tumor stage, the process may involve also extracutaneous sites, foremost lymph nodes and, less frequently, bone marrow and internal organs. The malignant T cells have a CD4+ CD8-phenotype with frequent loss of CD7 and to a lesser extent CD5. SS, a leukemic form of CTCL, is usually seen as a very aggressive disease with skin and blood involvement and a characteristic erythroderma.
Both MF and SS have no cure, and show clinical heterogeneity. Response to treatment is of limited duration and in advanced cases, characterized by recurrences, probably due to treatment resistance. It is also known that CTCL has been proposed to be a malignancy of three separate T cell populations: FOXP3+Treg, Th2 T cells and Th17 T cells [56-58].
But what is the etiology of MF and SS? The pathogenesis of the disease remains unclear and poorly understood. Studies have shown that no environmental triggers have been identified [59,60], infectious agents could aggravate the disease but there is no evidence that act as the cause [61], whereas more recent evidence shows a heterogeneous genetic landscape in CTCL [23,24,62-66]. Somatic genetic alterations were frequently found in genes involved in specific cellular processes and signaling pathways, including epigenetic regulation, DNA damage response, cell cycle control, programmed cell death, T Cell Receptor (TCR) signaling, as well as the nuclear factor-kappa B (NF- κB), MAPK signaling, Janus kinase (JAK)/signal transducer and activator of transcription (Stat) pathways [23,24,62-67].
Pathogenesis of the malignant phenotype assumes that underlying genetic mutations sensitize the pre-mycosis fungoides cells to become and stay activated, proliferate abnormally in response to stimulatory signals, and achieve clonal dominance with accumulation in the skin and subsequent lymph nodes, blood, and/or other organs. At this stage, the role of microenvironment in CTCL development plays a major and key role.
Microenvironment in CTCL
It has been determined that MF and SS arose from distinct T-cell subsets: MF from skin-resident effector memory T-cells (TRm) and SS from central memory T-cells (TCm) [68]. TRm cells were shown to express Cutaneous Lymphocyte-Associated Antigen (CLA) and CCR4, but lack L-selectin and CCR7 expression, which would enable them to access lymph nodes and circulation, while central memory T cells (TCm) express L-Selectin and CCR7 as well as CLA and CCR4, which enables them to affect, skin, lymph nodes and blood resulting in greater morbidity and mortality to the host [69]. Skin microenvironment plays an important role in the development and progression of mycosis fungoides/Sezary syndrome. Tumorinfiltrating T cells, regulatory T cells (Tregs), dendritic cells, macrophages, myeloid-derived suppressor cells, and mast cells, all have a crucial role in the pathogenesis of mycosis fungoides/Sezary syndrome [70-73]. The interaction between these cells and the skin microenvironment is the key in the proliferation of neoplastic T cells and their escape from immunosurveillance [74-76].
Chemokine receptor expression on tumor cells of MF differs according to disease stage. Therefore, in patch and plaque stage (early stage), epidermal keratinocytes and dermal fibroblasts express CXCL9/CXCL10 and induce chemotaxis and recruitment of CXCR3- positive T cells (tumor cells) and the microenvironment consists of non-malignant Th1 cells and CD8+ tumor-infiltrating T-cells. A significant proportion of the immune cells are activated CD8+ T cells and T helper 1 (Th1) cells expressing cytotoxic molecules, implying that early inflammation encompasses a cell-mediated anti-tumor response that actively suppresses the expansion of the malignant cells [77-79]. It is also known that in early-stage MF, Signal Transducers and Activators of Transcription (STAT) 4, the activation of which is required for Th1 differentiation, is overexpressed by IL-12 signaling via JAK2/ TYK2. In early stage MF, the skin has shown normal to increased expression of IFNγ, IL-12 and IL-2. Furthermore, it has been demonstrated that while normal T cells express b1 and b2 chains of the IL-12 receptor, CTCL malignant Th2 cells only express IL-12R b1 chain and thus are not sensitive even to exogenous IL-12 [80]. STAT5 can be activated via IL-2, IL-7 and IL-15 signaling (in the early disease stages) or via constitutively active in malignant cells JAK1 and JAK3 signaling (cytokine-independent signaling) [81,82] as described in Figure 2. STAT5 upregulates the expression of cell survival genes (Bcl-2 and BCL-x), cell cycle genes (Cyclin D and c-Myc), Th2 cytokines (IL-4) and miR-155 [81-84]. Inhibition of this protein signaling in CTCL results in inhibition of proliferation [81]. Advanced stages, such as tumor MF stage and SS, are considered Th2- type diseases (Figure 2). The Th2-dominant microenvironment is advantageous for tumor cells, because Interferon (IFN)-γ- producing Th1 cells enhance immune responses against the tumor. Langerhans cells and dermal fibroblasts express CCL17 which induces chemotaxis and recruitment of CCR4-positive T cells. Moreover, CCL17 is expressed by Langerhans cells and endothelial cells in lesional skin of MF and SS [85,86] suggesting important roles of CCL17–CCR4 interaction in preferential traffic of tumor cells of MF and SS to the skin. Epidermal keratinocytes express CCL27 which induces chemotaxis and recruitment of CCR10-positive T cells. At this stage of disease, IL-2 and IL-15 signaling via JAK1 and JAK3 kinases activates STAT5, which increases the expression of oncogenic miR- 155 [83] and subsequently inhibits STAT4 expression [87] resulting in a switch from Th1 to Th2 phenotype in malignant T cells. During this switch, the expression of STAT6 is often upregulated in CTCL [86].
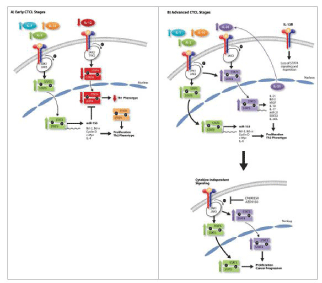
Figure 2: STAT signaling in Cutaneous T-cell lymphoma in early and in advance stage (Adapted from Netchiporouk et al, 2014).
Disease progression, and therefore the Th1 to Th2 switch, can occur due to dysfunctional apoptosis due to decreased and/or defective FAS death receptor expression in neoplastic T-cells has been associated with advanced disease and was recognized as an immune escape mechanism in CTCL [58,90]. Malignant T-cells in MF and SS overexpress IL-2 receptor subunit alpha (IL2RA; CD25) that results from a cascade of phosphorylation of several proteins including JAK3, STAT3 and STAT5 [96,97]. Querfeld et al. have provided evidence for T-cell exhaustion as an immunosuppressive mechanism in CTCL [91]. The chronic production of Th2 cytokines, such as IL-4, IL-5, and IL-10 by the malignant T cell population likely represents one mechanism by which the tumor cells circumvent the antitumor immune response. As the malignancy progresses to late stage MF, there have shown a loss of Th1 cytokines with a concomitant increase in the expression of other cytokines, such as IL-4, IL-5, IL-10 and IL-13. In the advanced stages, such constitutive STAT3 activation, which increases survival and resistance to apoptosis and promotes Th2 and Th17 phenotypes, is induced by an IL-21 autocrine signaling loop [92] the presence of IL-7 and IL-15 in the microenvironment [82], and/or constitutive cytokine-independent activation of JAK1 and JAK3 signaling [93,94].
Examination of clonal T cells in skin and blood samples of mycosis fungoides/Sezary syndrome patients has shown that they commonly express activation markers such as CD25 (IL-2R) or CD45RO. Guenova et al (2013) [95] in a previous study demonstrated that culture of benign T cells away from the malignant clone reduced Th2 (IL4 and GATA3) and enhanced Th1 responses (IFNγ and t-BET) but separate culture had no effect on malignant T cells. Co-culture of healthy T cells with L-CTCL T cells reduced IFNγ production and neutralizing antibodies to IL-4 and IL-13 restored Th1 responses. T cells isolated from the skin lesions of patients with L-CTCL had similar responses; IFNγ increased in non-clonal T cells after separate co-culture but remained unchanged in malignant T cells. Additionally, information from Dimitroff and his colleagues [96] has shown that clonal T cells in L-CTCL patients expressed a distinctive Th2 signature characterized by elevated expression of IL-4 and IL-13, whereas found increased levels of IL-10 in clonal malignant CD4 cells and in CD8 T cells of L-CTCL patients compared with healthy controls. Clark et al. observed that isolated T cells from either peripheral blood or skin lesions of CTCL patients contained a population of cells with high forward and side scatter characteristics on flow cytometric analysis [97]. A similar population of High-Scatter T cells (THS) was not observed in samples obtained from patients with benign conditions. More importantly, these high-scatter T cells, upon careful immunophenotyping and analysis of clonal TCRVb chain expression, were convincingly shown to represent the malignant T cell clone.
Pharmaceutical JAK/STAT inhibition current treatment
The conventional current treatment of CTCL includes skindirected therapies, radiation therapy, systemic therapy which includes biologic or immune therapies (bexarotene, INFγ, ECP), chemotherapy (methotrexate, doxorubicin, gemcitabine), Histone Deacetylase (HDAC) inhibitors (vorinostat and romidepsin). In the therapeutic field, there are many treatments under investigation including molecules such as a) immune checkpoint inhibitors, which are antibodies that target immune checkpoints (anti-PD-1 and anti-PD-L1), b) monoclonal antibodies such as CD30, an example of which is brentuximab which targets and binds to tumor cells with CD30 expression, which then leads to tumor cell killing, FDA approved for. Hodgkin lymphoma and systemic anaplastic large cell lymphoma, c) proteasome inhibitors, such as bortezomib which is approved to treat patients with multiple myeloma or mantle cell lymphoma, is being evaluated in clinical trials in combination with an HDAC inhibitor (eg, vorinostat, romidepsin), chemotherapies, or other targeted agents.
In the past, our research team with our collaborators from Bioinformatics had tried to identify the top 1000 diffentially expressed genes in MF and normal samples using two GEO datasets (p value ‹0.01). This analysis ended up with 623 upregulated genes and 377 down regulated genes implicated in various biological pathways. The differentially expressed genes formed disease signatures that were queries in a well-established drug repurposing pipeline (LINCS-1000). From this effort, the top 20 drugs were collected for which we found their structures in Chem Spider database for each gene list with the most negative enrichment scores suggesting that the drugs were acting as inhibitors (unpublished data). Two of the compounds that draw our attention were bortezomib and fedraditinib. Our work on CTCL cell lines and bortezomid indicated that combinational therapy with bortezomib/methotrexate had an inferior impact on the apoptosis of CTCL compared to monotherapy, with bortezomib presenting as the most efficient treatment option for SS and methotrexate for MF [98]. Additionally, both agents, but mostly bortezomib, significantly deregulate a large number of genes in SS and MF cell lines, suggesting another pathway through which these agents could induce apoptosis in CTCL. Finally, we show that SS and MF respond differently to treatment, verifying their distinct nature and further emphasizing the need for discrete treatment approaches [98].
On the other hand, fedratinib is a JAK2 inhibitor which has been used in trials studying the treatment and basic science of Solid Tumor, Myelofibrosis, Renal Impairment, Neoplasm Malignant, and Hepatic Impairment, among others. Sanofi, with an announcement on November 18, 2013, stated the discontinuation of the investigational development of Fedratinib, but Celgene recently has re-started fedratinib development. Classical Hodgkin lymphoma and primary mediastinal large B-cell lymphoma with 9p24.1/JAK2 copy gain(s) were sensitive to treatment with the JAK2-selective inhibitor fedratinib both in vitro and in vivo [99]. It would be of our interest to investigate the effects of fedratinib in CTCL alone or in combination with other synergistic compounds.
There is scientific evidence were treatment with ruxolitinib (JAK1/2 inhibitor) inhibited CTCL cell proliferation and JAK/STAT activity in CTCL cell lines, while activated apoptosis and inhibited DNA synthesis in CTCL cells [100]. Other evidence demonstrated that combination of ruxolitinib with vorinostat (HDAC inhibitor) had synergistic effects against hematological disease [101]. Kopp et al. document that miR-155 is a novel downstream target of STAT5 and is involved in malignant proliferation of T cells and he demonstrated that treatment of malignant cells with JAK inhibitor tofacitinib (CP 690550) strongly inhibits miR-155 expression and STAT5 activation [83].
Conclusion
The above results suggest a potential therapeutic role of JAK inhibitors based on the promising results from in vitro studies. Our research interest is that the above mentioned compounds have been already used successfully for a variety of neoplasmatic diseases, apart from in vitro studies. Therefore, it would be very interesting and of great importance to investigate the role of these inhibitors either alone or in combination (different Jak inhibitors, or Jak inhibitors with other such as HDAC inhibitors) in in vitro studies in CTCL in order to evaluate their role as a therapeutic strategy for this disease, followed by examination of their role and efficacy in in vivo models as well as in human patients with an end goal the designing of clinical trials. This knowledge has as a main target for such drugs to be used effectively in CTCL therapy providing the best benefit for the patient. Moreover, the above studies could also lead to the development of dual or multidrug inhibitors with reduced cytotoxicity, resistance and/or side effects for patients with CTCL.
References
- Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009; 228: 273-287.
- Seavey MM, Dobrzanski P. The many faces of Janus kinase. Biochem Pharmacol. 2012; 83: 1136-1145.
- Kisseleva T, Bhattacharya S, Braunstein J, Schindler C. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002; 285: 124.
- Kim HP, Imbert J, Leonard WJ. Both integrated and differential regulation of components of the IL-2/ IL-2 receptor system. Cytokine Growth Factor Rev. 2006; 17: 349-366.
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008; 133: 775-787.
- D’Souza WN, Lefrancois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. 2003; 171: 5727-5735.
- Holgate ST, Polosa R. Treatment strategies for allergy and asthma. Nat Rev Immunol. 2008; 8: 218-230.
- Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008; 9: 1341-1346.
- Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol. 2008; 9: 1347-1355.
- Hauber HP, Bergeron C, Hamid Q. IL-9 in allergic inflammation. Int Arch Allergy Immunol. 2004; 134: 79-87.
- Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008; 29: 848-862.
- Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008; 26: 57-79.
- Bhattacharya S, Braunstein J, Schindler C. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002; 285: 124.
- Kawamura M, McVicar DW, Johnston JA, Blake TB, Chen YQ, Lal BK, et al. Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes. Proc Natl Acad Sci USA. 1994; 91: 6374-6378.
- Musso T, Johnston JA, Linnekin D, Varesio L, Rowe TK, O’Shea JJ, et al. Regulation of JAK3 expression in human monocytes: phosphorylation in response to interleukins 2, 4, and 7. J Exp Med. 1995; 181: 1425-1431.
- Tortolani PJ, Lal BK, Riva A, Johnston JA, Chen YQ, Reaman GH, et al. Regulation of JAK3 expression and activation in human B cells and B cell malignancies. J Immunol. 1995; 155: 5220-5226.
- Gurniak CB, Berg LJ. Murine JAK3 is preferentially expressed in hematopoietic tissues and lymphocyte precursor cells. Blood. 1996; 87: 3151-3160.
- Verbsky JW, Bach EA, Fang YF, Yang L, Randolph DA, Fields LE. Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells. J Biol Chem. 1996; 271: 13976-13980.
- O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012; 36: 542-550.
- Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011; 34: 39-49.
- Liao W, Schones DE, Oh J, Cui Y, Cui K, Roh TY, et al. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nat Immunol. 2008; 9: 1288-1296.
- Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain-of-function alteration in RLTPR (p.Q575E). Blood. 2017; 130: 1430-1440.
- da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, et al. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet. 2015; 47: 1465-1470.
- McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015; 126: 508-519.
- Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J Leukoc Biol. 2018; 104: 499-514.
- Furumoto Y, Gadina M. The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. Bio Drugs. 2013; 27: 431-438.
- Bao L, Zhang H, Chan LS. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. JAKSTAT. 2013; 2: e24137.
- Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs. 2017; 77: 521-546.
- Kapoor R, Menon C, Hoffstad O, Bilker W, Leclerc P, Margolis DJ. The prevalence of atopic triad in children with physician-confirmed atopic dermatitis. J Am Acad Dermatol. 2008; 58: 68-73.
- Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis--part II: immune cell subsets and therapeutic concepts. J Allergy Clin Immunol. 2011; 127: 1420-1432.
- Chapoval S, Dasgupta P, Dorsey NJ, Keegan AD. Regulation of the T helper cell type 2 (Th2)/T regulatory cell (Treg) balance by IL-4 and STAT6. J Leukoc Biol. 2010; 87: 1011-1018.
- Alves de Medeiros AK, Speeckaert R, Desmet E, Van Gele M, De Schepper S, Lambert J. JAK3 as an Emerging Target for Topical Treatment of Inflammatory Skin Diseases. PLoS One. 2016; 11: e0164080.
- Esaki H, Ewald DA, Ungar B, Rozenblit M, Zheng X, Xu H, et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J Allergy Clin Immunol. 2015; 135: 153-163.
- Tanimoto A, Shinozaki Y, Yamamoto Y, Katsuda Y, Taniai-Riya E, Toyoda K, et al. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: Comparison with conventional therapeutic agents. Exp Dermatol. 2018; 27: 22-29.
- Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. 2015; 136: 667-677.
- Rodrigues MA. Torres T JAK/STAT inhibitors for the treatment of atopic dermatitis. J Dermatolog Treat. 2019; 1-20.
- Boehncke WH, Scho¨ MP. Psoriasis. Lancet. 2015; 386: 983-994.
- Wohn C, Ober-Blöbaum JL, Haak S, Pantelyushin S, Cheong C, Zahner SP, et al. Langerin(neg) conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc Natl Acad Sci USA. 2013; 110: 10723-10728.
- Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Muñoz-Planillo R, Hasegawa M, et al. Staphylococcus d -toxin induces allergic skin disease by activating mast cells. Nature. 2013; 503: 397-401.
- Van Dyken SJ, Mohapatra A, Nussbaum JC, Molofsky AB, Thornton EE, Ziegler SF, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and γs d T cells. Immunity. 2014; 40: 414-424.
- Ghoreschi K, Thomas P, Breit S, Dugas M, Mailhammer R, van Eden W, et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003; 9: 40-46.
- Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol. 2014; 134: 2757-2767.
- Cochez PM, Michiels C, Hendrickx E, Van Belle AB, Lemaire MM, Dauguet N, et al. AhR modulates the IL-22-producing cell proliferation/recruitment in imiquimod-induced psoriasis mouse model. Eur J Immunol. 2016; 46: 1449- 1459.
- Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. 2005; 11: 43-49.
- Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014; 32: 227-255.
- Papp K, Menter M, Abe M, Elewski B, Feldman SR, Gottlieb AB, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015; 173: 949-961.
- Feldman SR, Thac¸iD, Gooderham M, Augustin M, de la Cruz C, Mallbris L, et al. Tofacitinib improves pruritus and health-related quality of life up to 52 weeks: results from 2 randomized phase III trials in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol. 2016; 75: e31162-1170.
- Bachelez H, van de Kerkhof PC, Strohal R, Kubanov A, Valenzuela F, Lee JH, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet. 2015; 386: 552-561.
- Papp KA, Menter MA, Raman M, Disch D, Schlichting DE, Gaich C, et al. A randomized phase 2b trial of baricitinib, an oral Janus kinase (JAK) 1/JAK2 inhibitor, in patients with moderate-to-severe psoriasis. Br J Dermatol. 2016; 174: 1266-1276.
- Ports WC, Khan S, Lan S, Lamba M, Bolduc C, Bissonnette R, Papp K. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor to facitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013; 169: 137-145.
- Punwani N, Burn T, Scherle P, Flores R, Shi J, Collier P, et al. Downmodulation of key inflammatory cell markers with a topical Janus kinase 1/2 inhibitor. Br J Dermatol. 2015; 173: 989-997.
- Dummer R. Future perspectives in the treatment of Cutaneous T-Cell Lymphoma (CTCL). Semin Oncol. 2006; 33: S33-S36.
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHOEORTC classification for cutaneous lymphomas. Blood. 2005; 105: 3768- 3785.
- Querfeld C, Zain J, Rosen ST. Primary Cutaneous T-Cell Lymphomas: Mycosis Fungoides and Sezary Syndrome. Cancer Treat Res. 2019; 176: 225-248.
- Scarisbrick JJ, Quaglino P, Prince HM, Papadavid E, Hodak E, Bagot M, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2018.
- Berger CL, Tigelaar R, Cohen J, Mariwalla K, Trinh J, Wang N, et al. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood. 2005; 105: 1640-1647.
- Dummer R, Heald PW, Nestle FO, Ludwig E, Laine E, Hemmi S, et al. Sezary syndrome T-cell clones display T-helper 2 cytokines and express the accessory factor-1 (interferon-gamma receptor beta-chain). Blood. 1996; 88: 1383-1389.
- Krejsgaard T, Ralfkiaer U, Clasen-Linde E, Eriksen KW, Kopp KL, Bonefeld CM, et al. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J Invest Dermatol. 2011; 131: 1331-1338.
- Litvinov IV, Tetzlaff MT, Rahme E, Jennings MA, Risser DR, Gangar P, et al. Demographic patterns of cutaneous T-cell lymphoma incidence in Texas based on two different cancer registries. Cancer Med. 2015; 4: 1440-1447.
- Litvinov IV, Tetzlaff MT, Rahme E, Jennings MA, Risser DR, Gangar P, et al. Identification of geographic clustering and regions spared by cutaneous T-cell lymphoma in Texas using 2 distinct cancer registries. Cancer. 2015; 121: 1993-2003.
- Willerslev-Olsen A, Krejsgaard T, Lindahl LM, Bonefeld CM, Wasik MA, Koralov SB, et al. Bacterial toxins fuel disease progression in cutaneous T-cell lymphoma. Toxins (Basel). 2013; 5: 1402-1421.
- Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015; 47: 1011-1019.
- Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic analysis of mycosis fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nat Genet. 2015; 47: 1056-1060.
- Kiel MJ, Sahasrabuddhe AA, Rolland DC, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrentmutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun. 2015; 6: 8470.
- Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet. 2015; 47: 1426-1434.
- Woollard WJ, Pullabhatla V, Lorenc A, et al. Candidate driver genes in Sezary syndrome: frequent perturbations of genes involved in genome maintenance and DNA repair. Blood. 2016; 127: 3387-3397.
- Bastidas Torres AN, Najidh S, Tensen CP, Vermeer MH. Molecular advances in cutaneous T-cell lymphoma. Semin Cutan Med Surg. 2018; 37: 81-86.
- Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010; 116: 767-771.
- Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. 2015; 7: 269rv1.
- Rabenhorst A, Schlaak M, Heukamp LC, Förster A, Theurich S, von Bergwelt-Baildon M, et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood. 2012; 120: 2042-2054.
- Wu X, Schulte BC, Zhou Y, Haribhai D, Mackinnon AC, Plaza JA, et al. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J Invest Dermatol. 2014; 134: 2814- 2822.
- Sugaya M, Miyagaki T, Ohmatsu H, Suga H, Kai H, Kamata M, et al. Association of the numbers of CD163(ω) cells in lesional skin and serum levels of soluble CD163 with disease progression of cutaneous T cell lymphoma. J Dermatol Sci. 2012; 68: 45-51.
- Vermeer MH, van Doorn R, Dukers D, Bekkenk MW, Meijer CJ, Willemze R. CD8+ T cells in cutaneous T-cell lymphoma: expression of cytotoxic proteins, Fas Ligand, and killing inhibitory receptors and their relationship with clinical behavior. J Clin Oncol. 2001; 19: 4322-4329.
- Jawed SI, Myskowski PL, Horwitz S, Moskowitz A, Querfeld C, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part I. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014; 70: 205.
- Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015; 13: 45.
- Miyagaki T, Sugaya M. Immunological milieu in mycosis fungoides and Sezary syndrome. J Dermatol. 2014; 41: 11-18.
- Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, et al. Immunopathogenesis and therapy of cutaneous Tcell lymphoma. J Clin Invest. 2005; 115: 798-81
- Wood GS, Edinger A, Hoppe RT, Warnke RA. Mycosis fungoides skin lesions contain CD8+ tumor-infiltrating lymphocytes expressing an activated, MHC-restricted cytotoxic T-lymphocyte phenotype. J Cutan Pathol. 1994; 21: 151-156.
- Hsi AC, Lee SJ, Rosman IS, Carson KR, Kelley A, Viele V, et al. Expression of helper T cell master regulators in inflammatory dermatoses and primary cutaneous T-cell lymphomas: diagnostic implications. J Am Acad Dermatol. 2015; 72: 159-167.
- Showe LC, Fox FE, Williams D, Au K, Niu Z, Rook AH. Depressed IL-12- mediated signal transduction in T cells from patients with Sezary syndrome is associated with the absence of IL-12 receptor beta 2 mRNA and highly reduced levels of STAT4. J Immunol. 1999; 163: 4073-4079.
- Dobbeling U. Transcription factor profiling shows new ways towards new treatment options of cutaneous T cell lymphomas. Curr Drug Discov Technol. 2007; 4: 24-30.
- Qin JZ, Kamarashev J, Zhang CL, Dummer R, Burg G, Dobbeling U. Constitutive and interleukin-7- and interleukin-15-stimulated DNA binding of STAT and novel factors in cutaneous T cell lymphoma cells. J Invest Dermatol. 2001; 117: 583-589.
- Kopp KL, Ralfkiaer U, Gjerdrum LM, Helvad R, Pedersen IH, Litman T, et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle. 2013; 12: 1939-1947.
- Litvinov IV, Pehr K, Sasseville D. Connecting the dots in Cutaneous T Cell Lymphoma (CTCL): STAT5 regulates malignant T cell proliferation via miR- 155. Cell Cycle. 2013; 12: 2172-2173.
- Kakinuma T, Sugaya M, Nakamura K, Kaneko F, Wakugawa M, Matsushima K, et al. Thymus and activationregulated chemokine (TARC/CCL17) in mycosis fungoides: serum TARC levels reflect the disease activity of mycosis fungoides. J Am Acad Dermatol. 2003; 48: 23-30.
- Ferenczi K, Fuhlbrigge RC, Pinkus J, Pinkus GS, Kupper TS. Increased CCR4 expression in cutaneous T cell lymphoma. J Invest Dermatol. 2002; 119: 1405-1410.
- Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D, Duvic M. Deregulation in STAT signaling is important for Cutaneous T-Cell Lymphoma (CTCL) pathogenesis and cancer progression. Cell Cycle. 2014; 13: 3331- 3335.
- Dereure O, Levi E, Vonderheid EC, Kadin ME. Infrequent Fas mutations but no Bax or p53 mutations in early mycosis fungoides: a possible mechanism for the accumulation of malignant T lymphocytes in the skin. J Invest Dermatol. 2002; 118: 949-956.
- Wu J, Nihal M, Siddiqui J, Vonderheid EC, Wood GS. Low FAS/CD95 expression by CTCL correlates with reduced sensitivity to apoptosis that can be restored by FAS upregulation. J Invest Dermatol. 2009; 129: 1165-1173.
- Sommer VH, Clemmensen OJ, Nielsen O, Wasik M, Lovato P, Brender C, et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia. 2004; 18: 1288-1295.
- Querfeld C, Leung S, Myskowski PL, Curran SA, Goldman DA, Heller G, et al. Primary T cells from cutaneous T-cell lymphoma skin explants display an exhausted immune checkpoint profile. Cancer Immunol Res. 2018; 6: 900-909.
- van der Fits L, Out-Luiting JJ, van Leeuwen MA, Samsom JN, Willemze R, Tensen CP, et al. Autocrine IL-21 stimulation is involved in the maintenance of constitutive STAT3 activation in Sezary syndrome. J Invest Dermatol. 2012; 132: 440-447.
- Nielsen M, Kaestel CG, Eriksen KW, Woetmann A, Stokkedal T, Kaltoft K, et al. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/ Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia. 1999; 13: 735-738.
- van Kester MS, Out-Luiting JJ, von dem Borne PA, Willemze R, Tensen CP, Vermeer MH. Cucurbitacin I inhibits Stat3 and induces apoptosis in Sezary cells. J Invest Dermatol. 2008; 128: 1691-1695.
- Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res. 2013; 19: 3755-3763.
- Cedeno-Laurent F, Watanabe R, Teague JE, Kupper TS, Clark RA, Dimitroff CJ. Galectin-1 inhibits the viability, proliferation, and Th1 cytokine production of nonmalignant T cells in patients with leukemic cutaneous T-cell lymphoma Blood. 2012; 119: 3534-3538.
- Clark RA, Shackelton JB, Watanabe R, Calarese A, Yamanaka K, Campbell JJ, et al. High-scatter T cells: a reliable biomarker for malignant T cells in cutaneous T-cell lymphoma. Blood. 2011; 117: 1966-1976.
- Mpakou V, Papadavid E, Kontsioti F, Konsta E, Vikentiou M, Spathis A, et al. ‘Apoptosis Induction and Gene Expression Profile Alterations of Cutaneous T-Cell Lymphoma Cells following Their Exposure to Bortezomib and Methotrexate.’, PLoS One. 2017; 12: e0170186.
- Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res. 2014; 20: 2674-2683.
- Pérez C, González-Rincón J, Onaindia A, Almaráz C, García-Díaz N, Pisonero H, et al. Mutated JAK kinases and deregulated STAT activity are potential therapeutic targets in cutaneous T-cell lymphoma, Haematologica. 2015; 100: e450-e453.
- Civallero M, Cosenza M, Pozzi S, Sacchi S. Ruxolitinib combined with vorinostat suppresses tumor growth and alters metabolic phenotype in hematological diseases. Oncotarget. 2017; 8: 103797-103814.