
Case Report
Ann Hematol Oncol. 2019; 6(6): 1251.
Primary Isolated Amyloidoma of Head and Neck: Case Reports and Literature Review
Ciavarella S1*, Quinto AM1, Negri A1, Opinto G1, Sciacovelli AM1, Lotito MR2, Fedrigo M3, Castellani C3 and Guarini A1
1Hematology and Cell Therapy Unit, IRCCS - Istituto Tumori “Giovanni Paolo II”, Italy
2Radiology and Imaging Unit, IRCCS - Istituto Tumori “Giovanni Paolo II”, Italy
3Department of Cardiac Thoracic and Vascular Sciences, University of Padua, Italy
*Corresponding author: Ciavarella S, Hematology and Cell Therapy Unit, IRCCS – Istituto Tumori “G. Paolo II” Viale O. Flacco, 65 - 70125 - Bari, Italy
Received: March 06, 2019; Accepted: April 26, 2019;Published: May 03, 2019
Abstract
Primary Isolated Amyloidomas (PIA) represent rare localized deposits of amyloid, which can occur at different body sites including the central nervous system and neck structures. Only single case reports and sporadic case series have described clinical, pathological and molecular features of head and neck PIA, which share some aspects concerning diagnostic methodologies, prognostic criteria and treatment strategies. Here, we describe two cases of PIA localized within brain and larynx, respectively, and analyze the diagnostic role of different clinical, imaging and histopathological examinations. Due to their rarity, the etiology and pathogenesis of head and neck PIA remain poorly understood, and our report aims at enriching and reviewing the literature in this specific field.
Keywords: Amyloidosis; Primary isolated amyloidoma; Head and neck amyloidoma
Case Presentation
The term “Primary Isolated Amyloidoma” (PIA) refers to a solitary amyloid-containing lesion that may localize at different sites, including musculoskeletal, respiratory, gastrointestinal systems and, rarely, head and neck compartment [1]. Classically, PIA is not accompanied neither by primary or secondary amyloid-producing systemic conditions as multiple myeloma or systemic amyloidosis, nor increased risk for their development.
Extracellular accumulation of insoluble, fibrillar polypeptides, namely amyloid, has the unique property of resisting to enzymatic digestion, facilitating continuous deposition within tissues. Although mostly composed by immunoglobulin-derived kappa or lambda chains, the etiology of PIA formation remains unknown. Even more little is known about head and neck PIA, which can involve brain, cranial nerves, spinal neural structures, nasopharynx, larynx, and parapharyngeal spaces [2]. Cerebral deposition of amyloid, for instance, may accompany many inflammatory or degenerative disorders of Central Nervous System (CNS) or simulate cerebral lymphomas, astrocytoma, and other inflammatory processes. Similarly, pharynx-laryngeal localization may simulate chronic inflammatory polyp or other indolent neo-formations. In absence of specific laboratory signs, these entities can be diagnosed only by accurate instrumental evaluation, including Magnetic Resonance Imaging (MRI), Computerized Tomography (CT) and, more recently, 18-Fluorodeoxyglucose (FDG)-based Positron-Emission Tomography/CT (PET/CT) as well as histomorphologic and immunochemical electron microscopy to identify the amyloid subtype [3]. However, based on their exceptional localization and anecdotal description, uncertainty exists about PIA management, especially for those with unusual head and neck localizations.
Here, we report two clinical cases of PIA with common biological features despite different localization within the head and neck region, and elucidate key aspects of diagnosis, prognosis and treatment.
Case 1
History: A 51-year old woman was admitted at our Center complaining a bilateral continuous tinnitus occurred many years before. She reported a recent episode of absence epilepsy with no additional neurological symptoms. She underwent neurological examination revealing no suggestive signs and was thus prescribed for neuroimaging with head CT and MRI. Blood cell count and serology screens (electrolytes, kidney and liver function tests, acute inflammatory protein dosage, serum and urine protein electrophoresis plus immunofixation, kappa/lambda light chain ratio, and beta2-microglobulin) resulted within normal ranges. A bone marrow biopsy showed no marrow infiltration by clonal plasma cells.
Imaging assessment: The non-contrast head CT revealed an irregular mass with apparent intra-axial extension and prevalent localization within the left temporal area. The lesion showed weak hyper-density and surrounded by moderate edema. Early mass effect and cerebral convolution flattening were also visible. Contrastenhanced MRI demonstrated the supratentorial, deep temporal lesion appearing hypo-intense on T2-weighted imaging and enhancing moderately with gadolinium (Figure 1A). Moreover, spectroscopy indicated a significant increase of choline peak and presence of lipids and lactates. The radiologic differential diagnosis ranged between lymphoproliferative disorders, malignant neoplastic primaries, secondary tumor localizations, and inflammatory granulomatosis processes. A total body CT excluded neoplastic primaries, systemic lymphadenopathy and other organ alterations. Based on these results, the patient underwent a wide surgical excision of the lesion (via left temporal craniotomy) with diagnostic purpose.
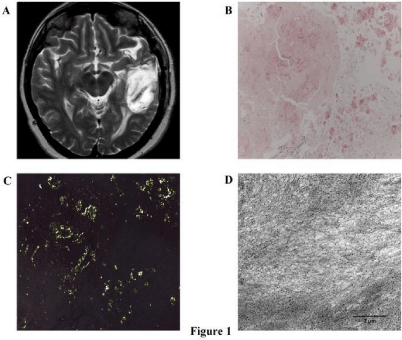
Figure 1: Radiological and electron microscopy pictures of cerebral PIA.
(A) Representative axial MRI scan showing a 4.6x2.9 cm left temporal
expansive mass with inhomogeneous contrast enhancement. (B, C) The Red
Congo staining defines the amyloidotic deposit in the biopsy tissue (200x
of magnification) with classical bi-refringence under polarized light (200x of
magnification). (D) Representative transmission electron microscopy image
showing an inner portion of the lesion with positive staining for lambda-lightchain
fibrils of amyloid (9000X original direct magnification).
Histological and immunochemical findings: Histopatological examination of biopsy specimens revealed large deposition of amorphous material with variable grade of vascularization and infiltrated by scarce, mature CD38+ plasma cells clonally restricted for the immunohistochemical expression of immunoglobulin lambda light chain. Moreover, additional staining for CD20, BCL-1, GFAP and OLIG2 were negative, suggesting no further cellular infiltrates of both lymphoid and neuronal origin. The Congo-red staining was consistent with amyloid showing typical bi-refringence under polarized light (Figure 1B-C). Ultrastructural examination confirmed the presence of large amount of lambda light chain-positive amyloid fibrils within the lesion (Figure 1D). Finally, the histological evaluation of a bone marrow biopsy revealed only a 4% polyclonal plasma cell infiltration, thus definitely excluding an underlying systemic amyloidosis.
Treatment and follow-up: Surgical eradication of the isolated cerebral lesion represented, at the same time, the first diagnostic and treatment approach. The patient was then administered with anticonvulsive therapy and followed-up by MRI as long as 13 months. Laboratory tests and physical examination as well as the last imaging demonstrated no signs of relapse of progression to systemic disease.
Case 2
History: A 62-year old man presented with 5-year history of change in voice with no other symptoms. He had undergone a videoassisted fibro-laryngoscopy revealing bilateral not homogeneous edema of false vocal cords along with a nodular thickening of the left cord. At presentation, physical examination was completely normal as well as blood and serological picture including thyroid, kidney and liver functionality tests, protein electrophoresis, immunofixation and urine screening. Also chest x-ray and ultrasonography of the neck and echocardiography were within the limits. A total body CT excluded additional solid primaries.
Neuroimaging: The patients underwent head and neck MRI demonstrating a significant increase in volume of both vocal cords with no contrast enhancement (Figure 2A). Neither morphologic alteration of cartilaginous structure nor neck lymphadenopathy was noticed. Based on the scarce specificity of CT imaging and evidence of an apparent nodular lesion in previous laryngoscopy, a biopsy of the left cord was planned for diagnostic purpose.
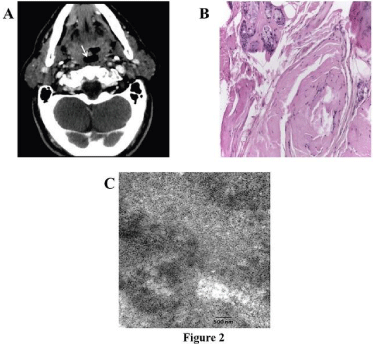
Figure 2: Radiological and electron microscopy pictures of laryngeal PIA.
(A) MRI scan of the larynx showing homogenous thickening of the false vocal
cord region, more evident at right (white arrow), with no significant areas of
contrast enhancement. (B) Ematoxillin and Eosin staining showed a small
deposit of amyloid within interstitium and around the submucosal glands
(200x of magnification). (C) Representative electron microscopy image of
positive staining for lambda-light-chain amyloid fibrils infiltrating the biopsy
specimen (9000X original direct magnification).
Histological findings: Histological examination described localized deposition of non-cellular eosinphilic material highly suggestive for amyloid. No plasma cell or lymphocyte infiltration was evident and Congo-red staining confirmed the diagnosis of PIA. Due to the exiguity of the biopsy specimen, immunohistochemistry resulted not reliable (Figure 2B), while only electron microscopy showed the infiltration of lambda light chain-positive amyloid fibrils (Figure 2C). A bone marrow biopsy showed no clonal plasma cell infiltration.
Treatment and Follow-Up
The patient remained in clinical and imaging (CT and RMI) follow-up for 12 months, with no need of specific therapeutic approach. Neither worsening of dysphonia nor occurrence of new symptoms was noticed during follow-up, thus supporting a watchfulwaiting strategy.
Discussion/Conclusions
This work was aimed at enriching the small series of case reports describing rare head and neck PIA. Roughly 40 cases of cerebral amyloidoma were reported, mostly focusing on neuroimaging or neurosurgical aspects (Table 1). In our case of cerebral PIA neither pathognomonic symptoms nor systemic abnormalities were detectable. Electron microscopy demonstrated lambda-restricted clonal plasma cells along with a pattern of AL-lambda chain deposition. In line with the higher prevalence of front-temporal brain PIA, we observed a supratentorial, deep temporal lesion in the white matter, showing a weak contrast enhancement in T2-weighted MRI images and scarce perifocal edema. In the absence of PET/CT scans, our imaging was insufficient to discern the mass nature. As reported, common neuroimaging lacks specificity for PIA, although it provides topographic insights guiding the surgical biopsy, essential to exclude primary CNS malignancies. In contrast, specific imaging patterns of small, periventricular and/or perivascular brain lesions may be suggestive of Alzheimer Disease (AD)-associated amyloid depositions [4]. PET/CT with radiotracers other than 18-FDG, as 18F-florbetapir and 18F-flutemetamol, were recently proven to detect amyloid-mediated abnormalities in vivo [5], distinguishing AD patterns from different disorders, including PIA. However, additional effort is needed to validate diagnostic and prognostic criteria to be routinely applied in the clinical practice.
![]()
Author
Symptoms
Tumor localization
Imaging
Treatment
Follow up
Harris, 1979
Seizure, headache
Frontal cortical and white matter
CT: calcific.
Surgery
NA
Radio-isotope (+)
Angiography
Spaar, 1981
Visual loss, headache, depression
Occipital, frontal white matter, touching ventricle
CT: hypodense
Surgery
3 months
Angiography
Linke, 1992
Visual disturbance, paraparesis both legs
Frontal, white matter, adjacent to ventricle
NA
Autopsy
NA
Cohen, 1992
Seizure, headache, cognitive dysfunction
1 cortical, 2 white matter
CT: CE
Surgery
2 years,
MRI: T1 hypointense
MRI : no progress
Erikson, 1993
Seizure, died after hemorrhage
Parietal, white matter, touching the ventricle
Scintigraphy (+)
Surgery/Autopsy
15 years
Angiography
Schroeder, 1995
Ataxia, leg weakness, cognitive decline
Parieto-occipital, white matter, ventricle
CT: hyperdense, CE
Sterotactical biopsy
41 months
Angiography
Laeng, 1998
Case 1: mental decline
Choroid plexus bilaterally
CT: CE
Autopsy
NA
Case 2: right hemisyndrome
Fronto-parietal, extending to the ventricle
CT: hyperdense, CE
Surgery
NA
MRI: CE, hypointense
Case 3: seizure
Frontal, near the ventricle
MRI: CE,
Sterotactical biopsy
NA
Angiography: avascular
Case 4: mental decline, numbness lower extremity
Bilateral, multifocal, parieto-occipital, periventricular
MRI: CE
Sterotactical biopsy
1 year
Smadja, 2000
Seizure
Frontal and temporal, white matter
CT: CE,
Sterotactical biopsy
NA
MRI:CE
Blatter, 2001
Hand paresis, dysarthria
Parieto-occipital, white matter, periventricular
MRI:CE
Sterotactical biopsy
5 years, MRI: slight progression
Gandhi, 2003
Seizure
Frontal, white matter, touching the ventricle
CT: hyperdense, calcifiction, CE
Surgery
NA
MRI: CE, T1 hypointense, T2 hypointense
Tabatabai, 2005
Mnestic decline
Subependymal, tegmentum, pons, capsular externa bilateral
MRI: CE, T1 hyperintense
Sterotactical biopsy
NA
FDG-PET
Meir, 2005
Hearing loss
Temporal
CT (native), MRI : CE, FDG-PET (+)
Sterotactical biopsy
18 months, MRI : doubled in size
Fischer, case
Seizure, leg paresis
Cortical/
CT: hyperdense, CE
Biopsy
4 years, MRI : slight progression
subcortical
MRI: CE, T1 hypointense, SPECT (+), FDG-PET (-)
Heb, 2017
Case 1:
Left occipital
MRI: minor CE
Biopsy
17 months
Confusion, headache, vomiting and visual deficit of the lower right quadrant, dyslexia
Case 2: Intermittent headache, right leg paresis
Left pre-central
CT and MRI: CEand minor perifocal edema
Biopsy
180 months
Case 3:
Right frontal
MRI: inhomogeneous CE
Gross total resection
11 months
Seizures
Case 4:
Right parieto – occipital
PET – MRI: inhomogeneous CE
Partial resection
24 months
Seizures, left homonymous hemianopsia, hemiparesis and numbness of left hand and foot
Case 5:
Right temporal
CT and MRI: CE, focal calcification and minor perifocal edema
Partial resection
71 months
Seizures
Case 6:
Left frontal
CT and MRI: inhomogeneous CE, midline shift, perifocal edema
Biopsy
21 months
Depression, frontal lobe disorder, seizures, numbness of the right half of the body
Case 7:
Right thalamus
MRI: thalamic lesion with midline shift
Partial resection
4 months
Vertigo
Table 1: List of recorded cases of isolated cerebral and laryngeal AL amyloidoma.
Also for laryngeal amyloidomas, imaging is not specific. In our case, the lesion appeared as a heterogeneous mass with low contrast enhancement, requiring a direct biopsy. This may be challenging in case of diffuse rather than nodular laryngeal involvement. Histologically, amyloid mostly accumulates as solitary masses ranging from few millimeters to some centimeters. Hematoxylin/Eosin staining commonly reveals eosinophilic material appearing yellowgreen bi-refringent under polarized light after Congo-red staining. Lymphocyte and macrophage aggregates may surround the principal mass and CD138+ plasma cells can be scattered within the fibrillar deposition. A typical lambda chain restriction can be demonstrated by immunohistochemistry, chromogenic in situ hybridization or, at RNA level, by IgH gene clonality assay. In our cases, cerebral and laryngeal PIA showed different immunochemical patterns, since the brain lesion was endowed with lambda-restricted plasma cells while the laryngeal one lacked any kind of cellular infiltration. However, the ultra-structural study characterized the nature of deposited amyloidogenic protein supporting this technique as the most specific one to investigate PIA and routine diagnostic activity in this field should be centralized into dedicated Centers.
In retrospective series, treatment strategies consist in surgery, adjuvant radiotherapy or steroids, depending on lesion localization, size and dimensional changes over time. Head and neck PIA share common initial approaches including surgical excision for diagnostic rather than treatment purpose. Complete excision is not always mandatory and, due to the benign nature of amyloid deposition, it is guided by the occurrence of peculiar signs or symptoms and their severity. In absence of stringent guidelines, neurosurgical approaches for cerebral PIA as well as microsurgical excision of laryngeal amyloidomas require accurate individualization. No evidence exists about the impact of steroids or adjuvant radiotherapy on patient long-term survival, excluding their routinely use and confining radiotherapy to cases for which surgery is prohibitive. On the contrary, based on the frequent observation of local relapses (de novo or as progression of residual plasma cell-infiltrated amyloid masses) often needing surgical revision, a regular imaging and laboratory followup remains to date the most common clinical recommendation for patients with head and neck PIA.
Acknowledgement
The Authors are grateful to Dr. Mila Della Barbera for technical support in the realization of images, and Dr. Carla Minoia and Dr. Giacomo Loseto for the external revision of the manuscript.
References
- Biewend ML, Menke DM, Calamia KT. The spectrum of localized amyloidosis: a case series of 20 patients and review of the literature. Amyloid. 2006; 13: 135-142.
- Prause CA, Zhai Q, Weindling SM. Pharyngeal amyloidomas: Variable appearance on imaging. Neuroradiol J. 2017; 30: 235-239.
- Glaudemans AW, Slart RH, Noordzij W, Dierckx RA, Hazenberg BP. Utility of 18F-FDG PET(/CT) in patients with systemic and localized amyloidosis. Eur J Nucl Med Mol Imaging. 2013; 40: 1095-1101.
- Parmar H, Rath T, Castillo M, Gandhi D. Imaging of focal amyloid depositions in the head, neck, and spine: amyloidoma. AJNR Am J Neuroradiol. 2010; 31: 1165-1170.
- Mekinian A, Jaccard A, Soussan M, Launay D, Berthier S, Federici L, et al. 18F-FDG PET/CT in patients with amyloid light-chain amyloidosis: case-series and literature review. Amyloid. 2012; 19: 94-98.